UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-Q
(Mark One)
☒ QUARTERLY
REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the quarterly period ended September 30,
2024
or
☐ TRANSITION
REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from __________ to __________
Commission File Number: 001-36374
ACTINIUM PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | | 74-2963609 |
(State or Other Jurisdiction of
Incorporation or Organization) | | (I.R.S. Employer
Identification No.) |
| | | |
| 100 Park Ave., 23rd Floor New York, NY | | 10017 |
| (Address of Principal Executive Offices) | | (Zip Code) |
(646) 677-3870
(Registrant’s Telephone Number, Including
Area Code)
Securities registered pursuant to Section 12(b)
of the Act:
| Title of each class | | Trading Symbol | | Name of exchange on which registered |
| Common stock, par value $0.001 | | ATNM | | NYSE American |
Indicate by check mark whether
the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the
preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such
filing requirements for the past 90 days. ☒ Yes No ☐
Indicate by check mark whether
the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T
(§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit
such files). ☒ Yes No ☐
Indicate by check mark whether
the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging
growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting
company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | | Accelerated filer | ☐ | |
| Non-accelerated filer | ☒ | | Smaller reporting company | ☒ | |
| | | | Emerging growth company | ☐ | |
If an emerging growth company,
indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial
accounting standards, provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether
the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). ☐ Yes
☒ No
Indicate the number of shares outstanding of each of the issuer’s
classes of common stock, as of November 14, 2024: 31,195,891
Actinium Pharmaceuticals, Inc.
Table of Contents
INDEX
PART I - FINANCIAL INFORMATION
ITEM 1. UNAUDITED FINANCIAL STATEMENTS
The accompanying condensed
consolidated financial statements have been prepared by Actinium Pharmaceuticals, Inc., or the Company, and are unaudited. In the opinion
of management, all adjustments (which include only normal recurring adjustments) necessary to present fairly the financial position at
September 30, 2024 and December 31, 2023, and the results of operations and cash flows for the three months and nine months ended September
30, 2024 and 2023, respectively, have been made. Certain information and footnote disclosures normally included in financial statements
prepared in accordance with accounting principles generally accepted in the United States of America have been condensed or omitted. It
is suggested that these financial statements be read in conjunction with the financial statements and notes thereto included in the Company’s
audited financial statements for the year ended December 31, 2023 in the Company’s Annual Report on Form 10-K. The results of operations
for the three months and nine months ended September 30, 2024 are not necessarily indicative of the operating results for the full year.
Actinium Pharmaceuticals, Inc.
Condensed Consolidated Balance Sheets
(Unaudited)
(amounts in thousands, except share and per share
data)
| | |
September 30,
2024 | | |
December 31,
2023 | |
| | |
(Unaudited) | | |
| |
| Assets | |
| | |
| |
| Current Assets: | |
| | |
| |
| Cash and cash equivalents | |
$ | 78,660 | | |
$ | 76,677 | |
| Prepaid expenses and other current assets | |
| 860 | | |
| 1,586 | |
| Total Current Assets | |
| 79,520 | | |
| 78,263 | |
| | |
| | | |
| | |
| Property and equipment, net of accumulated depreciation of $844 and $694 | |
| 412 | | |
| 550 | |
| Restricted cash – long term | |
| 322 | | |
| 313 | |
| Operating leases right-of-use assets | |
| 1,838 | | |
| 2,289 | |
| Finance leases right-of-use assets | |
| 22 | | |
| 30 | |
| Total Assets | |
$ | 82,114 | | |
$ | 81,445 | |
| | |
| | | |
| | |
| Liabilities and Stockholders’ Equity | |
| | | |
| | |
| | |
| | | |
| | |
| Current Liabilities: | |
| | | |
| | |
| Accounts payable and accrued expenses | |
$ | 7,195 | | |
$ | 7,953 | |
| Operating leases current liability | |
| 549 | | |
| 530 | |
| Finance leases current liability | |
| 11 | | |
| 11 | |
| Total Current Liabilities | |
| 7,755 | | |
| 8,494 | |
| | |
| | | |
| | |
| Long-term license revenue deferred | |
| 35,000 | | |
| 35,000 | |
| Long-term operating lease obligations | |
| 1,139 | | |
| 1,553 | |
| Long-term finance lease obligations | |
| 12 | | |
| 19 | |
| Total Liabilities | |
| 43,906 | | |
| 45,066 | |
| | |
| | | |
| | |
| Commitments and contingencies | |
| | | |
| | |
| | |
| | | |
| | |
| Stockholders’ Equity: | |
| | | |
| | |
| Preferred stock, $0.001 par value; 50,000,000 shares authorized, 0 shares issued and outstanding | |
| - | | |
| - | |
| Common stock, $0.001 par value; 1,000,000,000 shares authorized; 31,195,891 and 27,634,213 shares issued and outstanding, respectively | |
| 31 | | |
| 28 | |
| Additional paid-in capital | |
| 407,351 | | |
| 373,934 | |
| Accumulated deficit | |
| (369,174 | ) | |
| (337,583 | ) |
| Total Stockholders’ Equity | |
| 38,208 | | |
| 36,379 | |
| | |
| | | |
| | |
| Total Liabilities and Stockholders’ Equity | |
$ | 82,114 | | |
$ | 81,445 | |
See accompanying notes to the condensed consolidated
financial statements.
Actinium Pharmaceuticals, Inc.
Condensed Consolidated Statements of Operations
(Unaudited)
(amounts in thousands, except share and per share
data)
| | |
For the
Three Months Ended
September 30, | | |
For the
Nine Months Ended
September 30, | |
| | |
2024 | | |
2023 | | |
2024 | | |
2023 | |
| Revenue | |
| | |
| | |
| | |
| |
| Revenue | |
$ | - | | |
$ | - | | |
$ | - | | |
$ | - | |
| Other revenue | |
| - | | |
| - | | |
| - | | |
| - | |
| Total revenue | |
| - | | |
| - | | |
| - | | |
| - | |
| | |
| | | |
| | | |
| | | |
| | |
| Operating expenses: | |
| | | |
| | | |
| | | |
| | |
| Research and development, net of reimbursements | |
| 9,774 | | |
| 11,622 | | |
| 25,234 | | |
| 30,552 | |
| General and administrative | |
| 2,827 | | |
| 2,729 | | |
| 9,382 | | |
| 11,025 | |
| Total operating expenses | |
| 12,601 | | |
| 14,351 | | |
| 34,616 | | |
| 41,577 | |
| | |
| | | |
| | | |
| | | |
| | |
| Loss from operations | |
| (12,601 | ) | |
| (14,351 | ) | |
| (34,616 | ) | |
| (41,577 | ) |
| | |
| | | |
| | | |
| | | |
| | |
| Other income: | |
| | | |
| | | |
| | | |
| | |
| Interest income – net | |
| 1,033 | | |
| 1,075 | | |
| 3,025 | | |
| 2,083 | |
| Total other income | |
| 1,033 | | |
| 1,075 | | |
| 3,025 | | |
| 2,083 | |
| | |
| | | |
| | | |
| | | |
| | |
| Net loss | |
$ | (11,568 | ) | |
$ | (13,276 | ) | |
$ | (31,591 | ) | |
$ | (39,494 | ) |
| | |
| | | |
| | | |
| | | |
| | |
| Net loss per common share – basic and diluted | |
$ | (0.37 | ) | |
$ | (0.49 | ) | |
$ | (1.06 | ) | |
$ | (1.50 | ) |
| Weighted average common shares outstanding – basic and diluted | |
| 31,071,297 | | |
| 27,058,397 | | |
| 29,692,001 | | |
| 26,329,298 | |
See accompanying notes to the condensed consolidated
financial statements.
Actinium Pharmaceuticals, Inc.
Condensed Consolidated Statements of Changes
in Stockholders’ Equity
For the Period from January 1, 2024 to September
30, 2024
(Unaudited)
(amounts in thousands, except share amounts)
| | |
Common Stock | | |
Additional
Paid-In | | |
Accumulated | | |
Stockholders’ | |
| | |
Shares | | |
Amount | | |
Capital | | |
Deficit | | |
Equity | |
| Balance, January 1, 2024 | |
| 27,634,213 | | |
$ | 28 | | |
$ | 373,934 | | |
$ | (337,583 | ) | |
$ | 36,379 | |
| | |
| | | |
| | | |
| | | |
| | | |
| | |
| Stock-based compensation | |
| - | | |
| - | | |
| 1,378 | | |
| - | | |
| 1,378 | |
| Sale of common stock, net of offering costs | |
| 1,752,050 | | |
| 1 | | |
| 14,694 | | |
| - | | |
| 14,695 | |
| Issuance of common stock from exercise of stock options | |
| 10,148 | | |
| - | | |
| 75 | | |
| - | | |
| 75 | |
| Net loss | |
| - | | |
| - | | |
| - | | |
| (8,670 | ) | |
| (8,670 | ) |
| Balance, March 31, 2024 | |
| 29,396,411 | | |
$ | 29 | | |
$ | 390,081 | | |
$ | (346,253 | ) | |
$ | 43,857 | |
| | |
| | | |
| | | |
| | | |
| | | |
| | |
| Stock-based compensation | |
| - | | |
| - | | |
| 1,374 | | |
| - | | |
| 1,374 | |
| Sale of common stock, net of offering costs | |
| 1,154,191 | | |
| 2 | | |
| 9,955 | | |
| - | | |
| 9,957 | |
| Net loss | |
| - | | |
| - | | |
| - | | |
| (11,353 | ) | |
| (11,353 | ) |
| Balance, June 30, 2024 | |
| 30,550,602 | | |
$ | 31 | | |
$ | 401,410 | | |
$ | (357,606 | ) | |
$ | 43,835 | |
| | |
| | | |
| | | |
| | | |
| | | |
| | |
| Stock-based compensation | |
| 13,394 | | |
| - | | |
| 1,337 | | |
| - | | |
| 1,337 | |
| Sale of common stock, net of offering costs | |
| 631,895 | | |
| - | | |
| 4,604 | | |
| - | | |
| 4,604 | |
| Net loss | |
| - | | |
| - | | |
| - | | |
| (11,568 | ) | |
| (11,568 | ) |
| Balance, September 30, 2024 | |
| 31,195,891 | | |
$ | 31 | | |
$ | 407,351 | | |
$ | (369,174 | ) | |
$ | 38,208 | |
See accompanying notes to the condensed consolidated
financial statements.
Actinium Pharmaceuticals, Inc.
Condensed Consolidated Statements of Changes
in Stockholders’ Equity
For the Period from January 1, 2023 to September
30, 2023
(Unaudited)
(amounts in thousands, except share amounts)
| | |
Common Stock | | |
Additional
Paid-In | | |
Accumulated | | |
Stockholder’ | |
| | |
Shares | | |
Amount | | |
Capital | | |
Deficit | | |
Equity | |
| Balance, January 1, 2023 | |
| 25,674,823 | | |
$ | 26 | | |
$ | 355,220 | | |
$ | (288,765 | ) | |
$ | 66,481 | |
| | |
| | | |
| | | |
| | | |
| | | |
| | |
| Stock-based compensation | |
| - | | |
| - | | |
| 993 | | |
| - | | |
| 993 | |
| Sale of common stock, net of offering costs | |
| 54,414 | | |
| - | | |
| 770 | | |
| - | | |
| 770 | |
| Issuance of common stock from exercise of stock options | |
| 133 | | |
| - | | |
| 1 | | |
| - | | |
| 1 | |
| Net loss | |
| - | | |
| - | | |
| - | | |
| (11,037 | ) | |
| (11,037 | ) |
| Balance, March 31, 2023 | |
| 25,729,370 | | |
$ | 26 | | |
$ | 356,984 | | |
$ | (299,802 | ) | |
$ | 57,208 | |
| | |
| | | |
| | | |
| | | |
| | | |
| | |
| Stock-based compensation | |
| - | | |
| - | | |
| 1,001 | | |
| - | | |
| 1,001 | |
| Sale of common stock, net of offering costs | |
| 1,210,965 | | |
| 1 | | |
| 9,787 | | |
| - | | |
| 9,788 | |
| Issuance of common stock from exercise of stock options | |
| 44,108 | | |
| - | | |
| 261 | | |
| - | | |
| 261 | |
| Net loss | |
| - | | |
| - | | |
| - | | |
| (15,181 | ) | |
| (15,181 | ) |
| Balance, June 30, 2023 | |
| 26,984,443 | | |
$ | 27 | | |
$ | 368,033 | | |
$ | (314,983 | ) | |
$ | 53,077 | |
| | |
| | | |
| | | |
| | | |
| | | |
| | |
| Stock-based compensation | |
| 13,144 | | |
| - | | |
| 921 | | |
| - | | |
| 921 | |
| Sale of common stock, net of offering costs | |
| 415,854 | | |
| - | | |
| 2,861 | | |
| - | | |
| 2,861 | |
| Net loss | |
| - | | |
| - | | |
| - | | |
| (13,276 | ) | |
| (13,276 | ) |
| Balance, September 30, 2023 | |
| 27,413,441 | | |
$ | 27 | | |
$ | 371,815 | | |
$ | (328,259 | ) | |
$ | 43,583 | |
See accompanying notes to the condensed consolidated
financial statements.
Actinium Pharmaceuticals, Inc.
Condensed Consolidated Statements of Cash Flows
(Unaudited)
(amounts in thousands)
| | |
For the
Nine Months Ended
September 30, | |
| | |
2024 | | |
2023 | |
| Cash Flows Used In Operating Activities: | |
| | |
| |
| Net loss | |
$ | (31,591 | ) | |
$ | (39,494 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | |
| | | |
| | |
| Stock-based compensation expense | |
| 4,089 | | |
| 2,915 | |
| Depreciation & amortization expenses | |
| 607 | | |
| 591 | |
| Changes in operating assets and liabilities: | |
| | | |
| | |
| Prepaid expenses and other current assets | |
| 726 | | |
| (982 | ) |
| Accounts payable and accrued expenses | |
| (758 | ) | |
| (1,981 | ) |
| Operating lease right-of-use assets | |
| - | | |
| (527 | ) |
| Operating lease liabilities | |
| (394 | ) | |
| (367 | ) |
| Net Cash Used In Operating Activities | |
| (27,321 | ) | |
| (39,845 | ) |
| | |
| | | |
| | |
| Cash Flows Used In Investing Activities: | |
| | | |
| | |
| Purchase of property and equipment | |
| (11 | ) | |
| (153 | ) |
| Net Cash Used In Investing Activities | |
| (11 | ) | |
| (153 | ) |
| | |
| | | |
| | |
| Cash Flows Provided By Financing Activities: | |
| | | |
| | |
| Payments on finance leases | |
| (7 | ) | |
| (3 | ) |
| Sales of shares of common stock, net of costs | |
| 29,256 | | |
| 13,419 | |
| Proceeds from the exercise of stock options | |
| 75 | | |
| 262 | |
| Net Cash Provided By Financing Activities | |
| 29,324 | | |
| 13,678 | |
| | |
| | | |
| | |
| Net change in cash, cash equivalents, and restricted cash | |
| 1,992 | | |
| (26,320 | ) |
| Cash, cash equivalents, and restricted cash at beginning of period | |
| 76,990 | | |
| 109,608 | |
| Cash, cash equivalents, and restricted cash at end of period | |
$ | 78,982 | | |
$ | 83,288 | |
| | |
| | | |
| | |
| Supplemental disclosure of cash flow information: | |
| | | |
| | |
| Cash paid for interest | |
$ | - | | |
$ | - | |
| Cash paid for income taxes | |
$ | - | | |
$ | - | |
See accompanying notes to the condensed consolidated
financial statements.
Actinium Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(Unaudited)
Note 1 - Description of Business and Summary
of Significant Accounting Policies
Nature of Business -
Actinium Pharmaceuticals, Inc. is a biopharmaceutical company developing
Antibody Radiation Conjugates (“ARCs”) and other targeted radiotherapies to deliver cancer-killing radiation with cellular
level precision to treat patients with high unmet medical needs.
Basis of Presentation -
Unaudited Interim Financial Information - The accompanying unaudited interim condensed consolidated financial statements and related
notes have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”)
for interim financial information, and in accordance with the rules and regulations of the United States Securities and Exchange Commission
(the “SEC”) with respect to Form 10-Q and Article 10 of Regulation S-X. Accordingly, they do not include all of the information
and footnotes required by U.S. GAAP for complete financial statements. The unaudited interim condensed consolidated financial statements
furnished reflect all adjustments (consisting of normal recurring adjustments) which are, in the opinion of management, necessary for
a fair statement of the results for the interim periods presented. Interim results are not necessarily indicative of the results for the
full year. These unaudited interim condensed consolidated financial statements should be read in conjunction with the audited consolidated
financial statements and notes thereto contained in the Company’s Annual Report on Form 10-K for the year ended December 31, 2023.
Principles of Consolidation
- The basis of consolidation is unchanged from the disclosure in the Company’s Notes to the Consolidated Financial Statements
section in its Annual Report on Form 10-K for the year ended December 31, 2023. The unaudited condensed consolidated financial statements
include the Company’s accounts and those of the Company’s wholly owned subsidiaries.
Use of Estimates -
The preparation of these unaudited interim condensed consolidated financial statements in conformity with U.S. GAAP requires management
to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the unaudited interim condensed
consolidated financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those
estimates.
Cash, Cash Equivalents
and Restricted Cash - The Company considers all highly liquid accounts with original maturities of three months or less to be cash
equivalents. The Company holds most of its cash equivalents in a money market account comprised of US Treasury notes. Balances held by
the Company are typically in excess of Federal Deposit Insurance Corporation insured limits.
The following is a summary
of cash, cash equivalents and restricted cash at September 30, 2024 and December 31, 2023:
| (in thousands) | |
September 30,
2024 | | |
December 31,
2023 | |
| Cash and cash equivalents | |
$ | 78,660 | | |
$ | 76,677 | |
| Restricted cash – long-term | |
| 322 | | |
| 313 | |
| Cash, cash equivalents and restricted cash | |
$ | 78,982 | | |
$ | 76,990 | |
Restricted cash relates to
a certificate of deposit held as collateral for a letter of credit issued in connection with the Company’s lease of corporate office
space.
Leases – The
Company has an operating lease for corporate office space and a finance lease for office equipment located at the corporate office space.
Leases with an initial term of 12 months or less are not recorded on the balance sheet; lease expense for these leases is recognized on
a straight-line basis over the lease term.
Fair Value Measurement
- Fair value is defined as the price that would be received to sell an asset, or paid to transfer a liability, in an orderly transaction
between market participants. A fair value hierarchy has been established for valuation inputs that gives the highest priority to quoted
prices in active markets for identical assets or liabilities and the lowest priority to unobservable inputs.
Revenue Recognition -
The Company recognizes revenue in accordance with Accounting Standards Codification (ASC) Topic 606, Revenue From Contracts With Customers
(“ASC 606"). Under ASC 606, an entity recognizes revenue when its customer obtains control of promised goods or services,
in an amount that reflects the consideration that the entity expects to receive in exchange for those goods or services. To determine
revenue recognition for arrangements within the scope of ASC 606, the entity performs the following five steps: (i) identify the contract(s)
with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price, including variable
consideration, if any; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue as
the entity satisfies a performance obligation. The Company only applies the five-step model to contracts when it is probable that the
entity will collect the consideration to which it is entitled in exchange for the goods or services it transfers to the customer.
At contract inception, once
the contract is determined to be within the scope of ASC 606, the Company assesses whether the promised goods or services promised within
each contract are distinct and, therefore, represent a separate performance obligation. Goods and services that are determined not
to be distinct are combined with other promised goods and services until a distinct bundle is identified. In determining whether goods
or services are distinct, the Company evaluates certain criteria, including whether (i) the customer can benefit from the good or
service either on its own or together with other resources that are readily available to the customer (capable of being distinct) and
(ii) the good or service is separately identifiable from other goods or services in the contract (distinct in the context of the
contract).
The Company then determines
the transaction price, which is the amount of consideration it expects to be entitled from a customer in exchange for the promised goods
or services for each performance obligation and recognizes the associated revenue as each performance obligation is satisfied. The Company’s
estimate of the transaction price for each contract includes all variable consideration to which it expects to be entitled. Variable consideration
includes payments in the form of collaboration milestone payments. If an arrangement includes collaboration milestone payments, the Company
evaluates whether the milestones are considered probable of being reached and estimates the amount to be included in the transaction price
using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value
is included in the transaction price.
ASC 606 requires the Company
to allocate the arrangement consideration on a relative standalone selling price basis for each performance obligation after determining
the transaction price of the contract and identifying the performance obligations to which that amount should be allocated. The relative
standalone selling price is defined in the revenue standard as the price at which an entity would sell a promised good or service separately
to a customer. The Company then recognizes as revenue the amount of the transaction price that is allocated to the respective performance
obligation as each performance obligation is satisfied, either at a point in time or over time, and if over time, recognition is based
on the use of an output or input method.
Collaborative Arrangements
- The Company follows the accounting guidance for collaboration agreements with third parties, which requires that certain transactions
between the Company and collaborators be recorded in its consolidated statements of operations on either a gross basis or net basis, depending
on the characteristics of the collaborative relationship, and requires enhanced disclosure of collaborative relationships. The Company
evaluates its collaboration agreements for proper classification in its consolidated statements of operations based on the nature of the
underlying activity. When the Company has concluded that it has a customer relationship with one of its collaborators, the Company follows
the guidance of ASC 606.
Grant Revenue –
The Company has a grant from a government-sponsored entity for research and development related activities that provides for payments
for reimbursed costs, which included overhead and general and administrative costs as well as an administrative fee. The Company recognizes
revenue from grants as it performed services under this arrangement. Associated expenses are recognized when incurred as research and
development expense. Revenue and related expenses are presented gross in the consolidated statements of operations.
License Revenue –
The Company entered into a product licensing agreement whereby the Company allowed a third party to commercialize a certain product
in specified territories using the Company’s trademarks. The terms of this arrangement includes payment to the Company for a combination
of one or more of the following: upfront license fees; development, regulatory and sales-based milestone payments; and royalties on net
sales of licensed products. The Company uses its judgment to determine whether milestones or other variable consideration should be included
in the transaction price.
Upfront license fees:
If the license to the Company’s intellectual property is determined to be distinct from the other performance obligations identified
in the arrangement, the Company will recognize revenue from upfront license fees allocated to the license when the license is transferred
to the licensee and the licensee is able to use and benefit from the license. For licenses that are bundled with other promises, the Company
determines whether the combined performance obligation is satisfied over time or at a point in time.
Development, regulatory
or commercial milestone payments: At the inception of each arrangement that includes payments based on the achievement of certain
development, regulatory and sales-based or commercial events, the Company evaluates whether the milestones are considered probable of
being achieved and estimates the amount to be included in the transaction price using the most likely amount method. If it is probable
that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price. Milestone payments
that are not within the Company’s or the licensee’s control, such as regulatory approvals, are not considered probable of
being achieved until regulatory approval is received. At the end of each subsequent reporting period, the Company will re-evaluate the
probability of achieving such development and regulatory milestones and any related constraint, and if necessary, adjust the Company’s
estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis and recorded as part of license
revenue during the period of adjustment.
Sales-based milestone payments
and royalties: For arrangements that include sales-based royalties, including milestone payments based on the volume of sales, the
Company will determine whether the license is deemed to be the predominant item to which the royalties or sales-based milestones relate
and if such is the case, the Company will recognize revenue at the later of (i) when the related sales occur, or (ii) when the performance
obligation to which some or all of the royalty has been allocated has been satisfied (or partially satisfied).
Upfront payments and fees
may require deferral of revenue recognition to a future period until the Company performs its obligations under these arrangements or
when it is probable that a significant reversal in the amount of cumulative revenue recognized will not occur or when the uncertainty
associated with any variable consideration is subsequently resolved. Amounts payable to the Company are recorded as accounts receivable
when the Company’s right to consideration is unconditional.
Research and Development
Costs - Research and development costs are expensed as incurred. These costs include the costs of manufacturing drug product, the
costs of clinical trials, costs of employees and associated overhead, and depreciation and amortization costs related to facilities and
equipment. Research and development reimbursements are recorded by the Company as a reduction of research and development costs.
Share-Based Payments -
The Company estimates the fair value of each stock option award at the grant date by using the Black-Scholes option pricing model.
The fair value determined represents the cost for the award and is recognized over the vesting period during which an employee is required
to provide service in exchange for the award. The Company accounts for forfeitures of stock options as they occur.
Net Loss Per Common Share
- Basic loss per common share is computed by dividing the net loss available to common stockholders by the weighted average number
of shares of common stock outstanding during the reporting period. For periods of net loss, diluted loss per share is calculated similarly
to basic loss per share because the impact of all potential dilutive common shares is anti-dilutive. For the three and nine months ended
September 30, 2024 and 2023, the Company’s potentially dilutive shares, which include outstanding common stock options, restricted
stock units and warrants, have not been included in the computation of diluted net loss per share as the result would have been anti-dilutive.
| (in thousands) | |
September 30,
2024 | | |
September 30,
2023 | |
| Stock Options | |
| 5,216 | | |
| 3,304 | |
| Restricted Stock Units | |
| 300 | | |
| 305 | |
| Warrants | |
| 9 | | |
| 1,442 | |
| Total | |
| 5,525 | | |
| 5,051 | |
Recently Issued Accounting
Pronouncements – In December 2023, The Financial Accounting Standards Board, “FASB” issued Accounting Standards
Update “ASU” 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures, to enhance the transparency
and decision usefulness of income tax disclosures. The amendments in ASU 2023-09 provide improvements primarily related to the rate reconciliation
and income taxes paid information included in income tax disclosures. The Company would be required to disclose additional information
regarding reconciling items equal to or greater than five percent of the amount computed by multiplying pretax income (loss) by the applicable
statutory tax rate. Similarly, the Company would be required to disclose income taxes paid (net of refunds received) equal to or greater
than five percent of total income taxes paid (net of refunds received). The amendments in ASU 2023-09 are effective January 1, 2025, including
interim periods. Early adoption is permitted for annual financial statements that have not yet been issued or made available for issuance.
The Company will evaluate the impact of ASU 2023-09 on its financial statements.
In November 2023, FASB issued
ASU 2023-07, Segment Reporting (Topic 280), Improvements to Reportable Segment Disclosures, which provides improvements
to reportable segment disclosure requirements, primarily through enhanced disclosures around segment expenses. ASU 2023-07 requires the
Company to disclose significant segment expenses that are regularly provided to the chief operating decision maker (“CODM”)
and included within each reported measure of segment profit or loss. ASU 2023-07 also requires that the Company disclose an amount for
other segment items by reportable segment, a description of their composition and provide all annual disclosures about a reportable segment’s
profit or loss and assets pursuant to Topic 280 during interim periods. The Company must also disclose the CODM’s title and position,
as well as certain information around the measures used by the CODM and an explanation of how the CODM uses the reported measures in assessing
segment performance and deciding how to allocate resources. For public entities with a single reportable segment, the entity must provide
all the disclosures required pursuant to ASU 2023-07 and all existing segment disclosures under Topic 280. The amendments of ASU 2023-07
are effective for the Company for annual periods beginning January 1, 2024, and effective for interim periods beginning January 1, 2025.
The Company will adopt this standard, effective January 1, 2024, and report on it in the Company’s Annual Report on Form 10-K for
the year ended December 31, 2024. The Company will update all required disclosures pursuant to ASU 2023-07 at that time.
In October 2021, FASB issued
ASU 2021-08, Business Combinations (Topic 805), Account for Contract Assets and Contract Liabilities from Contracts with Customers,
which provides guidance on accounting for contract assets and contract liabilities acquired in a business combination in accordance with
ASC 606. To achieve this, an acquirer may assess how the acquiree applied ASC 606 to determine what to record for the acquired revenue
contracts. Generally, this should result in an acquirer recognizing and measuring the acquired contract assets and contract liabilities
consistent with how they were recognized and measured in the acquiree’s financial statements. The amendments of ASU 2021-08 were
effective January 1, 2023, including interim periods. The Company will evaluate the impact of ASU 2021-08 on any future business combinations
the Company may enter in the future.
Note 2 - Commitments and Contingencies
On June 15, 2012, the Company
entered into a license and sponsored research agreement with Fred Hutchinson Cancer Research Center (“FHCRC”) to build upon
previous and ongoing clinical trials with apamistamab (licensed antibody). FHCRC has completed both a Phase 1 and Phase 2 clinical trial
with apamistamab. The Company has been granted exclusive rights to the antibody and related master cell bank developed by FHCRC. A milestone
payment of $1 million will be due to FHCRC upon U.S. Food and Drug Administration (“FDA”) approval of the first drug utilizing
the licensed antibody. Upon commercial sale of the drug, royalty payments of 2% of net sales will be due to FHCRC.
Note 3 - Leases
The Company determines if
an arrangement is a lease at inception. This determination generally depends on whether the arrangement conveys to the Company the right
to control the use of a fixed asset for a period of time in exchange for consideration. Control of an underlying asset is conveyed to
the Company if the Company obtains the rights to direct the use of and to obtain substantially all of the economic benefits from using
the underlying asset. The Company has lease agreements which include lease and non-lease components, which the Company has elected to
account for as a single lease component for all classes of underlying assets. Lease expense for variable lease components are recognized
when the obligation is probable. The Company made an accounting policy election to exclude from balance sheet reporting those leases with
initial terms of 12 months or less.
Right-of-use assets and liabilities
are recognized at commencement date based on the present value of lease payments over the lease term. ASC 842 requires a lessee to discount
its unpaid lease payments using the interest rate implicit in the lease or, if that rate cannot be readily determined, its incremental
borrowing rate. As an implicit interest rate was not readily determinable in the Company’s leases, the incremental borrowing rate
was used based on the information available at commencement date in determining the present value of lease payments.
The lease term for all of
the Company’s leases includes the non-cancellable period of the lease plus any additional periods covered by either a Company option
to extend (or not to terminate) the lease that the Company is reasonably certain to exercise, or an option to extend (or not to terminate)
the lease controlled by the lessor. Options for lease renewals have been excluded from the lease term (and lease liability) for the majority
of the Company’s leases as the reasonably certain threshold is not met.
At September 30, 2024, the
Company has two leases which have been capitalized in accordance with ASC 842, one for corporate office space and one for office equipment.
The Company entered into a lease for corporate office space effective June 1, 2022. The lease has a term of 5 years 2 months, with an
expiration date of July 30, 2027 and current annual rent of $0.6 million. The Company is also responsible for certain other costs, such
as insurance, utilities and maintenance. During 2023, the Company spent $0.5 million in improvements at its corporate office space, which
has been included in the value of the operating right-to-use asset.
The components of lease expense are as follows:
| | |
Three months ended | | |
Nine months ended | |
| (in thousands) | |
September 30,
2024 | | |
September 30,
2023 | | |
September 30,
2024 | | |
September 30,
2023 | |
| Operating lease expense | |
$ | 173 | | |
$ | 173 | | |
$ | 519 | | |
$ | 518 | |
| | |
| | | |
| | | |
| | | |
| | |
| Finance lease cost | |
| | | |
| | | |
| | | |
| | |
| Amortization of right-to-use assets | |
$ | 3 | | |
$ | 1 | | |
$ | 8 | | |
$ | 3 | |
| Interest on lease liabilities | |
$ | - | | |
$ | - | | |
$ | 1 | | |
$ | - | |
| Total finance lease cost | |
$ | 3 | | |
$ | 1 | | |
$ | 9 | | |
$ | 3 | |
Supplemental cash flow information related to leases
are as follows:
Cash flow information:
| | |
Nine months ended | |
| (in thousands) | |
September 30,
2024 | | |
September 30,
2023 | |
| | |
| | |
| |
| Cash paid for amounts included in the measurement of lease liabilities: | |
| | |
| |
| Operating cash flow use from operating leases | |
$ | 462 | | |
$ | 453 | |
| Operating cash flow use from finance leases | |
$ | 8 | | |
$ | 3 | |
| Financing cash flow use from finance leases | |
$ | 7 | | |
$ | 3 | |
| | |
| | | |
| | |
| Non-cash activity: | |
| | | |
| | |
| | |
| | | |
| | |
| Right-of-use assets obtained in exchange for lease obligations: | |
| | | |
| | |
| Operating leases | |
$ | - | | |
$ | - | |
| Finance Leases | |
$ | - | | |
$ | - | |
Weighted average remaining lease terms are as follows
at September 30, 2024:
| Weighted average remaining lease term: | | | | |
| Operating leases | | | 2.8 years | |
| Finance Leases | | | 2.3 years | |
As the interest rate implicit
in the leases was not readily determinable at the time that the leases were evaluated, the Company used its incremental borrowing rate
based on the information available in determining the present value of lease payments. The Company’s incremental borrowing rate
was based on the term of the lease, the economic environment of the lease and reflects the rate the Company would have had to pay to borrow
on a secured basis. Below is information on the weighted average discount rates used at the time that the leases were evaluated:
| Weighted average discount rates: | |
| |
| Operating leases | |
| 4.8 | % |
| Finance Leases | |
| 6.2 | % |
Maturities of lease liabilities
are as follows:
(in thousands)
Year ending December 31, | |
Operating
Leases | | |
Finance
Leases | |
| 2024 (excluding nine months ended September 30, 2024) | |
| 156 | | |
| 3 | |
| 2025 | |
| 630 | | |
| 11 | |
| 2026 | |
| 643 | | |
| 11 | |
| 2027 | |
| 380 | | |
| - | |
| Total lease payments | |
$ | 1,809 | | |
$ | 25 | |
| Less imputed interest | |
| (121 | ) | |
| (2 | ) |
| Present value of lease liabilities | |
$ | 1,688 | | |
$ | 23 | |
Note 4 – Other revenue
The Company has a grant from
a government-sponsored entity for research and development related activities that provides payments for reimbursed costs, which included
overhead and general and administrative costs, as well as an administrative fee. The Company recognizes revenue from grants as it performed
services under this arrangement. Associated expenses are recognized when incurred as research and development expense. There was no grant
revenue recognized for the nine months ended September 30, 2024 and 2023, respectively.
On April 7, 2022, the Company
entered into a license and supply agreement (the “License Agreement”) with Immedica Pharma AB (“Immedica”), pursuant
to which Immedica licensed the exclusive product rights for commercialization of Iomab-B (I-131 apamistamab) in the European Economic
Area, Middle East and North Africa (“EUMENA”), including Algeria, Andorra, Bahrain, Cyprus, Egypt, Iran, Iraq, Israel, Jordan,
Kuwait, Lebanon, Libya, Monaco, Morocco, Oman, Palestine, Qatar, San Marino, Saudi Arabia, Switzerland, Syria, Tunisia, Turkey, the United
Arab Emirates, the United Kingdom, the Vatican City and Yemen. Upon signing, the Company was entitled to an upfront, non-refundable payment
of $35 million from Immedica, which was received in May 2022. Under the terms of the License Agreement, the Company is eligible to receive
certain regulatory and commercial milestone payments and royalties on net sales of the product in certain countries that may result from
the License Agreement. The Company continues to retain commercialization rights in the U.S. and rest of the world.
The Company’s contract
liabilities are recorded within Other revenue deferred – current liability or Long-term license revenue deferred in its interim
unaudited condensed consolidated balance sheets, depending on the short-term or long-term nature of the payments to be recognized. The
Company’s contract liabilities primarily consist of advanced payments from licensees. Long-term license revenue deferred was $35
million at September 30, 2024 and December 31, 2023; this deferred revenue will be recognized upon European Union’s regulatory approval
of Iomab-B or provision of definitive feedback that Iomab-B will not receive approval in the European Union.
Note 5 - Equity
In August 2020, the Company
entered into the Capital on Demand™ Sales Agreement with JonesTrading Institutional Services LLC, “JonesTrading”, pursuant
to which the Company may sell, from time to time, through or to JonesTrading, up to an aggregate of $200 million of its common stock.
On June 28, 2022, the Company entered into an Amended and Restated Capital on Demand™ Sales Agreement (the “A&R Sales
Agreement”) with JonesTrading and B. Riley Securities, Inc. (“B. Riley”). The A&R Sales Agreement modifies the original
Capital on Demand™ Sales Agreement to include B. Riley Securities as an additional sales agent thereunder. Shares of common stock
are offered pursuant to a shelf registration statement on Form S-3 (File No. 333-242322) filed with the SEC on August 7, 2020 (the “Prior
Shelf Registration Statement”). On August 11, 2023, the Company filed a new registration statement on Form S-3 (File No. 333-273911),
which registration statement was amended on February 2, 2024, and declared effective on February 5, 2024, to replace the Prior Shelf Registration
Statement, including a base prospectus which covers the offering, issuance and sale of up to $500 million of common stock, preferred stock,
warrants, units and/or subscription rights; and a sales agreement prospectus covering the offering, issuance and sale of up to a maximum
aggregate offering price of $200 million of common stock that may be issued and sold under the Amended Sales Agreement.
For the nine months ended
September 30, 2024, the Company sold 3.5 million shares of common stock, resulting in gross proceeds of $29.9 million and net proceeds
of $29.3 million. For the nine months ended September 30, 2023, the Company sold 1.7 million shares of common stock, resulting in gross
proceeds of $13.8 million and net proceeds of $13.4 million.
Stock Options
The following is a summary
of stock option activity for the nine months ended September 30, 2024:
| (in thousands, except for per-share amounts) | | Number of Shares | | | Weighted
Average
Exercise
Price ($) | | | Weighted
Average
Remaining
Contractual
Term
(in years) | | | Aggregate
Intrinsic
Value | |
| Outstanding, January 1, 2024 | | | 5,445 | | | $ | 6.80 | | | | 8.70 | | | $ | 373 | |
| Granted | | | 104 | | | | 7.28 | | | | | | | | | |
| Exercised | | | (10 | ) | | | 7.39 | | | | | | | | | |
| Cancelled | | | (323 | ) | | | 11.15 | | | | | | | | | |
| Outstanding, September 30, 2024 | | | 5,216 | | | | 6.54 | | | | 7.80 | | | | - | |
| | | | | | | | | | | | | | | | | |
| Exercisable, September 30, 2024 | | | 2,409 | | | | 8.09 | | | | 6.80 | | | | - | |
During the nine months ended
September 30, 2024, the Company granted new employees options to purchase 0.1 million shares of common stock with an exercise price ranging
from $7.20 to $8.15 per share, a term of 10 years, and a vesting period of 4 years. The options have an aggregated fair value of $0.5
million that was calculated using the Black-Scholes option-pricing model. Variables used in the Black-Scholes option-pricing model include:
(1) discount rate range from 4.11% to 4.5% (2) expected life of 6 years, (3) expected volatility range from 80.5% to 81.5%, and (4) zero
expected dividends.
During the nine months ended
September 30, 2024, options to purchase 323 thousand common shares were cancelled upon the termination of employment and 10 thousand options
were exercised for shares of common stock.
The fair values of all options
issued and outstanding are being amortized over their respective vesting periods. The unrecognized compensation expense at September 30,
2024 was $10.1 million related to unvested options, which is expected to be expensed over a weighted average of 3.0 years. During the
nine months ended September 30, 2024 and 2023, the Company recorded compensation expense related to stock options of $3.6 million and
$2.4 million, respectively.
Restricted Stock Units
The following is a summary
of restricted stock unit (“RSU”) activity for the nine months ended September 30, 2024:
| (in thousands, except for per-share amount) | |
RSUs | | |
Weighted
Average
Grant date Fair Value
Per Share ($) | |
| Outstanding, January 1, 2024 | |
| 305 | | |
| 5.89 | |
| Granted | |
| - | | |
| - | |
| Vested | |
| - | | |
| - | |
| Cancelled | |
| (5 | ) | |
| 8.31 | |
| Outstanding, September 30, 2024 | |
| 300 | | |
| 5.85 | |
The RSUs vest at the earliest
of a change of control event, the termination of the recipient’s continuous service status for any reason other than by the Company
for cause and the third anniversary of the date of the grant. The fair value of the RSUs, $1.8 million, was determined based on the stock
price on the dates of the grant and is being recognized over three years. The unrecognized compensation expense at September 30, 2024
of $0.5 million is expected to be expensed over 0.9 years. During the nine months ended September 30, 2024 and 2023, the Company recorded
compensation expense related to RSUs of $0.4 million in each reporting period.
Warrants
Following is a summary of
warrant activity for the nine months ended September 30, 2024:
| (in thousands, except for per-share amounts) | | Number of Shares | | | Weighted
Average
Exercise
Price ($) | | | Weighted
Average
Remaining
Contractual
Term
(in years) | | | Aggregate
Intrinsic
Value | |
| Outstanding, January 1, 2024 | | | 1,442 | | | $ | 16.42 | | | | 0.34 | | | $ | - | |
| Granted | | | - | | | | - | | | | | | | | | |
| Expired | | | (1,433 | ) | | | 16.13 | | | | | | | | | |
| Outstanding, September 30, 2024 | | | 9 | | | $ | 61.46 | | | | 3.48 | | | $ | - | |
| | | | | | | | | | | | | | | | | |
| Exercisable, September 30, 2024 | | | 9 | | | $ | 65.52 | | | | 3.09 | | | $ | - | |
ITEM 2. MANAGEMENT’S DISCUSSION AND ANALYSIS
OF FINANCIAL CONDITION AND RESULTS OF OPERATION
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING
STATEMENT NOTICE
This Quarterly Report on Form
10-Q and other reports filed by the Company from time to time with the Securities and Exchange Commission contains or may contain certain
forward-looking statements and information that are based upon beliefs of, and information currently available to, the Company’s
management as well as estimates and assumptions made by Company’s management. Readers are cautioned not to place undue reliance
on these forward-looking statements, which are only predictions and speak only as of the date hereof. For this purpose, any statements
contained in this Quarterly Report on Form 10-Q that are not statements of historical fact may be deemed to be forward-looking statements.
Without limiting the foregoing, words such as “may,” “will,” “expect,” “believe,”
“anticipate,” “estimate” or “continue” or comparable terminology are intended to identify forward-looking
statements. These statements by their nature involve substantial risks and uncertainties, and actual results may differ materially
depending on a variety of factors, many of which are not within our control. These factors include but are not limited to economic
conditions generally and in the industries in which we may participate; competition within our chosen industry, including competition
from much larger competitors; technological advances and failure to successfully develop business relationships. Although we believe that
the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance,
or achievements. Except as required by applicable law, including the securities laws of the United States, we do not intend to update
any of the forward-looking statements to conform these statements to actual results.
Description of Business
Actinium
Pharmaceuticals, Inc. (“Actinium”, the “Company”, or “we”) develops Antibody Radiation
Conjugates (“ARCs”) and other targeted radiotherapies intended to meaningfully improve outcomes for patients with
relapsed or refractory cancer who have failed existing therapies. We are deploying our technology platform, which we believe to be
industry-leading, and intellectual property with over 230 issued and pending patents worldwide, to develop ARCs and next-generation
radiotherapies against validated cancer targets.
Our Strategy
Actinium’s strategy
is to build an integrated, specialty radiotherapeutics company using the power of our platform technologies to deliver new treatment options
for patient populations living with high unmet medical needs in hematology and oncology. We believe our focus on relapsed or refractory
disease in cancer indications with high unmet medical need, with limited or no competition, and where the primary delivery of care occurs
in large comprehensive cancer care centers, is the appropriate strategy for our company. Radiotherapeutics leverage the cell-killing power
of linear energy transfer, which we believe is unmatched by other technologies and we also believe that relapsed or refractory disease
is an area where radiotherapeutics can succeed over other approaches. Our proven experience conducting multiple, late-stage clinical trials
provides an advantage operating a radiotherapy company where barriers related to their utilization and supply chain are higher than those
for other types of medicines due to the personalized, just-in-time nature of radiotherapies. We have accumulated extensive experience
coordinating the manufacturing and clinical supply chain of radiotherapeutics, resulting in the treatment of hundreds of patients across
diverse treatment settings, including the largest comprehensive cancer centers in North America, that we intend to leverage in our ongoing
and future development efforts. The potential of our approach is demonstrated by our product pipeline strategy, as well as the operating
model that we are building for our ARC candidates.
Our Product Candidate Pipeline
We have developed our lead
product candidates, Iomab-B and Actimab-A, to potentially improve outcomes for patients with relapsed or refractory acute myeloid leukemia
(“r/r AML”) to address the significant unmet need for better outcomes from treatment with therapeutics or from undergoing
a bone marrow transplant (“BMT”).
Our Next Generation ARC Pipeline
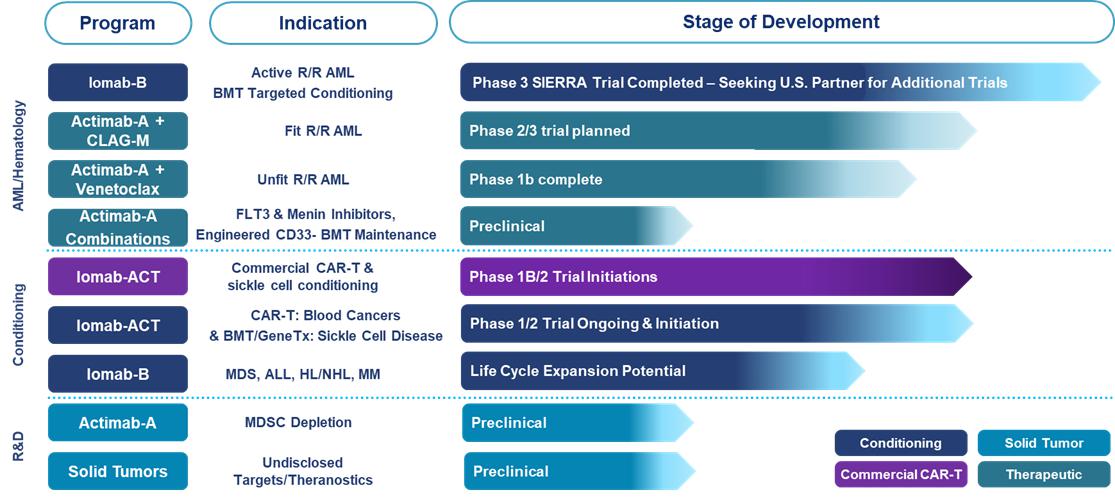
Iomab-B is a targeted radiotherapy
induction (disease control) and conditioning agent, comprised of the anti-CD45 monoclonal antibody apamistamab and the Iodine-131 (“I-131")
radioisotope payload, intended to enable patient access to BMT, the only potentially curative treatment option for patients with r/r AML.
Iomab-B has been studied in over 400 patients and is supported by data in six blood cancer indications, including a randomized Phase 3
trial in patients with r/r AML.
Actimab-A is being developed
as a targeted radiotherapeutic that uses the Actinium-225 (“Ac-225") isotope payload directed against CD33, a target expressed
ubiquitously in patients with AML and expressed in other myeloid malignancies. We are attempting to leverage the mutation-agnostic ability
of Ac-225 to establish Actimab-A as a backbone therapy in AML, an extremely heterogenous and radiosensitive disease, as a single agent
or in combinations with chemotherapy, targeted agents, cellular therapy and immunotherapy. Actimab-A has been studied in over 150 patients
and is being developed in collaboration with the National Cancer Institute (“NCI”) under a Cooperative Research and Development
Agreement (“CRADA”).
Iomab-ACT is our next-generation
CD45 targeted conditioning agent being developed for cell and gene therapies for both malignant and non-malignant hematologic indications.
We are developing Iomab-ACT with the goal of improving patient access and outcomes for potentially curative cell and gene therapies. Iomab-ACT
is currently being studied in three clinical trials, including with a commercial CAR-T therapy, prior to allogeneic BMT for patients with
sickle cell disease, which is intended to inform a trial design with gene therapy for sickle cell disease, and with a novel CD19 CAR-T
therapy.
We have active R&D efforts
leveraging our in-house preclinical and translational capabilities and are primarily focused on advancing several preclinical programs
for solid tumor indications.
Iomab-B
Iomab-B is a targeted radiotherapy
intended to provide both induction and conditioning in one agent. Iomab-B was evaluated in the randomized Study of Iomab-B in Elderly
Relapsed and Refractory AML (“SIERRA”) trial. The Phase 3 SIERRA trial enrolled 153 patients with r/r AML and compared outcomes
of patients receiving Iomab-B and a BMT for patients receiving physician’s choice of care with salvage chemotherapies and standard
allogeneic BMT in the control arm. The SIERRA trial was conducted in accordance with guidance from the End of Phase 2 meeting with the
FDA, which stated that positive results for durable Complete Remission (“dCR”) as the primary endpoint would be an acceptable
endpoint to support an Iomab-B Biologics License Application (“BLA”) filing. The Iomab-B dose in the SIERRA trial was also
individualized for each patient, based on the dosimetric infusion findings, to deliver 24 Gy radiation to the dose-limiting organ (typically
the liver), or 48 Gy to the bone marrow, whichever was predicted to receive the highest estimated dose of radiation.
In February 2023, Actinium
announced that the SIERRA trial met the primary endpoint with statistical significance, as 22% of patients (13/76) on the Iomab-B arm
achieved dCR compared to 0% of patients (0/77) on the control arm resulting in a p-value of <0.0001. The SIERRA trial did not, however,
meet the secondary endpoint in achieving a statistically significant improvement in overall survival in the intent to treat (“ITT”)
population.
On September 19, 2024, the
results of the SIERRA trial were published in the peer-reviewed Journal of Clinical Oncology, in an article titled, “Randomized
Phase III SIERRA Trial of 131I-Apamistamab Before Allogeneic Hematopoietic Cell Transplantation Versus Conventional Care
for Relapsed/Refractory Acute Myeloid Leukemia”.
Iomab-B Regulatory Status Update and Future Development
On August 5, 2024, Actinium
announced that it concluded both its clinical and Chemistry, Manufacturing and Controls (“CMC”) interactions with the FDA
regarding the BLA pathway for Iomab-B based on the SIERRA trial results. As previously disclosed, we had received positive feedback from
the FDA regarding our CMC package for Iomab-B and were also assigned a BLA number. However, in the third quarter of 2024, the FDA provided
definitive feedback that the SIERRA trial alone is not adequate to support a BLA filing for Iomab-B, despite (a) the SIERRA trial meeting
the primary endpoint of dCR with statistical significance (p-value<0.0001) and other positive secondary endpoints including Event Free
Survival (“EFS”) and safety, and (b) our presentation of several additional analyses from the SIERRA study, including long-term
follow-up demonstrating a trend towards improved overall survival and evidence of survival benefit in patients with high-risk TP53 mutations,
to support Iomab-B’s impact on overall survival. The FDA indicated that demonstrating an overall survival benefit in a randomized
head-to-head trial is necessary and has advised us to conduct a study to evaluate allogeneic BMT using Iomab-B plus a reduced intensity
conditioning regimen of fludarabine and total body irradiation (“Flu/TBI”) versus allogeneic BMT using reduced intensity conditioning
comprised of cyclophosphamide plus Flu/TBI. This proposed additional study differs from the SIERRA trial, which allowed physician’s
choice of salvage chemotherapies and heterogenous conditioning regimens in the control arm. Additionally, the proposed new study will
not allow patients to cross over from the control arm, which was allowed in the SIERRA trial and confounded the overall survival analysis
in the ITT patient population, as nearly 60% of patients crossed over from the control arm.
Actinium continued interactions
with the FDA in the third quarter of 2024 to further discuss the specifics of the additional head-to-head clinical trial required by the
FDA, including the patient population, which the FDA had suggested could include all adult AML patients. In the fourth quarter of 2024,
Actinium conducted a further meeting with the FDA. Based on this meeting, Actinium believes it has aligned with the FDA on the patient
population for a head-to-head Phase 3 clinical trial to further evaluate allogeneic BMT using Iomab-B plus a reduced intensity conditioning
regimen of Flu/TBI versus allogeneic BMT using reduced intensity conditioning comprised of cyclophosphamide plus Flu/TBI in all adult
patients aged 18 and above with active AML with blasts counts greater than 5% and less than 20%. This is a broader patient population
than the patients enrolled on the SIERRA trial, which only enrolled patients aged 55 and above. Further, the FDA now requires that an
additional dose optimization trial demonstrating safety and efficacy be completed to calculate the dose of Iomab-B based on absorbed dose
by the bone marrow, rather than the maximum tolerable dose of 24 Gy of radiation to the liver as was done in the SIERRA trial based on
several interactions we had with the FDA before starting the SIERRA trial. We are actively seeking a strategic partner for Iomab-B in
the U.S. to advance the head-to-head clinical trial or other clinical development activity for Iomab-B.
As previously disclosed and
noted above, Actinium also has a License Agreement with Immedica, granting Immedica the exclusive product rights for commercialization
of Iomab-B in certain countries in the European Economic Area, Middle East and North Africa (“EUMENA”) region.
Actimab-A
We have an industry-leading
clinical development program investigating our alpha-particle therapy Actimab-A, a CD33 targeting ARC conjugated to the potent alpha radiation
emitting isotope Ac-225. Actimab-A has been studied in over 150 patients across six clinical trials as both a single agent and in combination
with other modalities such as chemotherapy and targeted therapy. The potent linear energy transfer emitted by Ac-225 has no known resistance
mechanism and there is no known repair mechanism to the double strand DNA breaks it is capable of causing with a single alpha particle
hit to a cell. Actimab-A is currently being developed in combination with other regimens, including chemotherapies such as CLAG-M and
targeted agents such as venetoclax utilizing its potential mechanistic synergies. We are attempting to leverage the mutation-agnostic
ability of Ac-225 to establish Actimab-A as a backbone therapy in AML, an extremely heterogenous and radiosensitive disease.
In 2023, we entered into a
CRADA with NCI to develop Actimab-A for the treatment of patients with AML and other hematologic malignancies. The NCI will serve as the
regulatory sponsor for any clinical trials mutually approved by both parties to study Actimab-A, and the CRADA will provide extensive
support for and accelerate the development of Actimab-A alone or in combination with chemotherapy, immunotherapy, targeted agents and
other novel combinations. The CRADA studies will be overseen by the NCI in collaboration with Actinium’s clinical development team,
where we have the right to review and approve all protocols and have full rights to all data. The NCI CRADA provides for us to supply
Actimab-A and for NCI to cover all clinical trial execution and development expenses. The NCI CRADA is anticipated to have a balance sheet
sparing impact over the next several years. The NCI Cancer Therapy Evaluation Program (“CTEP”), which sponsors approximately
two thirds of all combination cancer studies, will accept Letters of Intent (“LOIs”) or concepts for Phase 1, 2 or 3 studies
of Actimab-A in AML and other hematological malignancies. To date, multiple Actimab-A study proposals have been submitted to the NCI,
which are being mutually reviewed and considered. We expect the NCI to initiate further development of Actimab-A in combination with CLAG-M
and other targeted agents to broaden the scope of its development in AML and other myeloid malignancies.
On October 23, 2024, the
NCI announced that its myeloMATCH program was officially open to patient enrollment across the U.S. and Canda. MyeloMATCH is a
portfolio of clinical trials to test precision medicine treatments for adults with AML or myelodysplastic syndrome
(“MDS”) being designed and led by four leading cancer research organizations including the Alliance for Clinical Trials
in Oncology, Canadian Cancer Trials Group, ECOG-ACRIN Cancer Research Group, and SWOG Cancer Research Network in collaboration with
the NCI National Clinical Trials Network (“NCTN”). Collectively, the myeloMATCH program expects to open trials at
hundreds of cancer care sites across the U.S. and Canada with the goal of enrolling 5,000 or more patients over the next several
years. Under our CRADA with the NCI, Actimab-A is part of the myeloMATCH program and may be included in future clinical trials.
Actimab-A Regulatory and Development Update
In the third quarter
of 2024, we met with the FDA to discuss the continued development of Actimab-A in combination with CLAG-M in patients with r/r AML. We
have aligned with the FDA on an operationally seamless randomized Phase 2/3 clinical trial design. Based on our interactions with the
FDA, this trial will first complete a Phase 2 portion where the Actimab-A dose will be optimized in combination with CLAG-M. Once the
optimized Actimab-A dose is determined, we expect the trial will seamlessly advance to the Phase 3 portion of the study, which is expected
to reduce time and resources required compared to separate Phase 2 and Phase 3 studies. We are also working to evaluate and develop multiple
clinical trials for Actimab-A across the treatment journey in AML and other myeloid indications, which could be conducted under the NCI
CRADA, investigator sponsored trials (“ISTs”) or Actinium’s sponsorship.
We continue to advance our
preclinical R&D efforts to further support the mutation agnostic profile of Actimab-A in AML and other CD33 expressing hematologic
malignancies as well as provide support for novel combinations to support its backbone therapy profile. In June 2024, we presented the
first-ever preclinical data demonstrating the combination of Actimab-A with leading menin inhibitors resulted in anti-tumor control and
potent leukemia cell killing in AML models at the 2024 European Hematology Association (“EHA”) Congress. We studied Actimab-A
in combination with the leading menin inhibitors, revumenib (Syndax Pharmaceuticals, Inc.) and ziftomenib (Kura Oncology, Inc.), which
are being developed for patients with KMT2A rearrangements and NMP1 mutations, which are present in approximately 10% and 30% of AML patients,
respectively. Actimab-A as a single agent showed potent in vitro AML cell killing activity in both MV-4-11 and MOLM-13 KMT2A mutant cell
lines, compared to the non-radio conjugated CD33 antibody lintuzumab (p<0.0001) and the combination of Actimab-A with leading menin
inhibitors triggered an acute increase in AML necrosis and cell death in vivo relative to single agent therapy within 72 hours of dosing.
Actimab-A enhanced AML cell death when combined with both revumenib and ziftomenib at all dose levels in difficult to treat KMT2A mutant
AML. Anti-tumor effect was significantly potentiated and prolonged when combining Actimab-A with a leading menin inhibitor compared to
monotherapies in xenograft leukemia models in vivo (p<0.0024 Actimab-A + menin). We have also evaluated Actimab-A in combination with
FLT3 (Fms-like tyrosine kinase 3) inhibitors such as gilteritinib (Astellas Pharma, Inc.) and midostaurin (Novartis Pharmaceuticals, Inc.).
FLT3 is one of the most commonly mutated genes in AML and is associated with aggressive disease with poor outcomes. Actimab-A was shown
to have single-agent activity against FLT3 mutant AML cell lines, supporting its mutation-agnostic mechanism, and enhanced the anti-leukemic
activity of the FLT3 inhibition in vitro.
We have also generated
preclinical data that depicts Actimab-A’s role in the tumor microenvironment to overcome immunosuppression driven by myeloid-derived
suppressor cells (“MDSCs”). We believe that our findings show that Actimab-A has the potential to selectively deplete MDSCs
in lung, colorectal and other cancers. Actimab-A also demonstrated statistically significant depletion of human MDSCs compared to Mylotarg®,
a CD33-targeted antibody-drug conjugate (“ADC”) in colorectal cancer (p<0.01), highlighting the potent cytotoxicity and
potential therapeutic benefit of radiotherapy compared to naked antibodies or ADCs.
Given the potentially large
addressable patient population for Actimab-A in both hematologic malignancies and solid tumors, we are open to pursuing strategic partnerships
or collaborations beyond our CRADA with the NCI that can further advance Actimab-A’s development for the benefit of patients.
Iomab-ACT
The opportunity exists for
better conditioning in other areas of cellular therapy, and we are working on a next generation conditioning program, Iomab-ACT, for the
rapidly growing cell and gene therapy market, as well as BMT conditioning for non-malignant hematologic indications such as sickle cell
disease. We are studying Iomab-ACT in collaboration with Memorial Sloan Kettering Cancer Center (“MSKCC”), for conditioning
prior to CAR-T therapy for patients with relapsed or refractory B-cell acute lymphoblastic leukemia (“B-ALL”) or diffuse large
B-cell lymphoma (“DLBCL”). This study funded by a NIH grant is the first study of its kind to use an ARC, with CAR-T therapy.
In October 2023, we announced the extension of a NIH Small Business Technology Transfer grant to support the clinical collaboration with
MSKCC. Most recently, at the 2024 Tandem Meetings | Transplantation & Cellular Therapy Meetings of ASTCT and CIBMTR, we presented
results from the ongoing Phase 1 trial. No patients (0/4) developed Immune Effector Cell-Associated Neurotoxicity Syndrome (“ICANS”)
of any grade, a major safety measure of the study, as ICANS is observed in 25% or more of patients with r/r B-ALL and DLBCL treated with
various CAR T-cell products and negligible incidence of cytokine release syndrome (“CRS”). Iomab-ACT demonstrated transient
depletion of peripheral blood lymphocytes and monocytes. Persistence of CAR T-cells up to 8 weeks and minimal non-hematologic toxicities
have been observed to date.
In May 2024, we announced
FDA clearance of an IND for a new clinical trial that will study Iomab-ACT as targeted conditioning prior to patients receiving an FDA
approved commercial CAR-T therapy. To our knowledge, this will be the first trial to study a targeted radiotherapy conditioning agent
with a commercial CAR-T therapy. Given the robust clinical data that exists with commercial CAR-T therapies, we believe this trial may
demonstrate the potential for Iomab-ACT to improve outcomes over current chemotherapy conditioning regimens, which we are seeking to replace
and provide patients better access to CAR-T. We expect to commence patient enrollment of this study in the first quarter of 2025 and generate
proof of concept clinical data by year end 2025.
In July 2024, we announced
a program for Iomab-ACT focused on providing patients with sickle cell disease broader access to cellular therapies including bone marrow
transplant and gene therapies. We also announced in July 2024 the FDA clearance of an IND for an investigator led clinical trial to study
Iomab-ACT as targeted conditioning prior to a BMT for patients with sickle cell disease in collaboration with Columbia University. Sickle
cell disease is a rare, debilitating and life-threatening blood disorder with significant unmet need that affects approximately 100,000
people in the U.S. Patients with sickle cell disease have a mutation that causes red blood cells to develop a crescent or “sickle”
shape, which restrict the flow in blood vessels and limit oxygen delivery to the body’s tissues, leading to severe pain and organ
damage called vaso-occlusive events (“VOEs”) or vaso-occlusive crises (“VOCs”). The recurrence of these events
or crises can lead to life-threatening disabilities and/or early death. A BMT is a potentially curative treatment option for patients
with sickle cell disease, particularly in pediatric patients who have had complications such as strokes, acute chest crises or recurring
pain crises due to their disease. We expect patient enrollment for the Phase 1 trial to commence in the first half of 2025 with initial
safety and efficacy results from the initial cohort of patients receiving Iomab-ACT prior to a BMT for their sickle cell disease beginning
in 2025. If successful, the trial is expected to inform a clinical trial to evaluate Iomab-ACT as a targeted conditioning agent prior
to gene therapy for which there are two approved agents for patients with sickle cell disease, Casgevy (Vertex Pharmaceuticals, Inc.)
and Lyfgenia (Bluebird Bio, Inc.).
The pipeline of CAR-T and
gene therapies has rapidly expanded, with the addressable patient population expected to nearly double and reach approximately 93,000
patients in the U.S. by 2030 based on the current pipeline of therapies. The CAR-T market size in terms of revenue is estimated to grow
at a CAGR of approximately 11% over the next five years. Currently, there are six CAR T-cell therapies approved by the FDA that are used
to treat patients with lymphomas, leukemia and multiple myeloma, which collectively had total sales over $3.5 billion in 2023. The addressable
market for Iomab-ACT is in line with the patient population for cellular therapy as all patients receive conditioning of some type prior
to these treatments. We will continue to develop Iomab-ACT based on early promising results, ultimately with the value proposition of
improving overall access and outcomes for patients who need cellular or gene therapies. We believe an opportunity exists for Iomab-ACT
to potentially generate significant revenue, if it can provide one or more clinical benefits related to lower CRS, less neurotoxicity,
longer duration of response or a higher overall success rate of cellular therapy due to benefits of targeted conditioning.
R&D and Preclinical ARC Programs
Our R&D capabilities have
the potential to yield differentiated, high-value ARC programs that demonstrate our experience across multiple validated cancer targets
and isotopes and cover broad areas of focus, leveraging our clinical development experience across hematology, targeted conditioning,
solid tumors, and next generation radiotherapies. We develop ARC product candidates that target antigens expressed on certain cancer cell
types and are able to destroy cellular DNA and kill these cells with the energy that they emit. The efficacy of ARCs does not require
internalization and stable linkers minimize off-target toxicity of the payload.
Our R&D programs inform
the advancement of our clinical programs. We have utilized our technology platform to develop our clinical portfolio in hematology –
Iomab-B and Actimab-A, in conditioning for transplant and as a therapeutic, respectively. Our differentiated R&D efforts are further
exemplified by our next-generation Iomab-ACT conditioning program for rapidly growing cell and gene therapies. Our platform has been used
to develop a pipeline of novel radiotherapeutic assets to drive company growth. We are working on several preclinical programs which include
novel approaches to validated cancer targets, as well as novel targets that we believe to show immense potential for radiotherapeutic
approaches. Preclinical pharmacology studies with our targeted radiotherapeutics, such as HER2, CD33 and CD38, have shown strong improvement
in tumor growth inhibition in various preclinical tumor models. These results have prompted our R&D team to spearhead expansion of
our efforts into multiple solid tumor programs in the preclinical stage with IND enabling studies underway. Leveraging our platform
and expertise in developing ARCs, we are exploring how nanobodies, single chain variable fragment (“scFv”), and other related
modalities can be combined with novel linkers and radioisotopes to enhance delivery to solid tumors.
We currently believe that
our ARCs are less likely than small molecules to face pricing pressure and negotiation from the Inflation Reduction Act of
2022 (“IRA”), given that small molecules are at risk for pricing negotiations seven years after approval compared to eleven
years for biologics with negotiated prices taking effect two years after selection. Further, a drug or biological product that has an
orphan drug designation, which Iomab-B and Actimab-A both have, for only one rare disease or condition will be excluded from the IRA’s
price negotiations requirements until such time the biological products has designations for more than one rare disease or condition,
or if is approved for an indication that is not within that single designated rare disease or condition, unless such additional designation
or such disqualifying approvals are withdrawn by the time CMS evaluates the drug for selection for negotiation. In addition, regulatory
barriers for a generic ARC are much higher than for small molecule radioligands such as those under development or approved, namely, Pluvicto®,
Lutathera®, and Xofigo®. While generic versions of certain radiopharmaceuticals utilizing peptides, which
are considered small molecules, have been submitted to the FDA via the ANDA pathway, ARCs fall under biologics. For this reason, only
the biosimilar approach pertains to ARCs filed under 351(k) BLA pathway. The regulatory pathway for biosimilar is much more comprehensive
than the pathway for generics, and it has not been proven that biosimilars are interchangeable with the innovator’s ARCs. In addition,
we are not aware of any regulations that would require us to provide Iomab-B or Actimab-A, including their respective monoclonal antibodies
(“mAbs”), apamistamab and lintuzumab, to any third party or potential competitor.
We seek to expand our capabilities
and technologies across therapeutic modalities, linker technologies and in vivo cancer models, and build visibility through presentations
at key conferences and publications in journals of high impact. Our R&D efforts are centered on the advancement of our key ARC programs
with a robust “fast-to-clinic” approach.
Our Platform and Ac-225 Manufacturing Technology
Our proprietary technology
platform is built on the core competency to produce targeted radiotherapeutics, and coupled with our know-how and intellectual property,
establishes our company in the development of isotope-agnostic, multi-targeted product candidates that have the potential to address the
treatment of hard-to-treat diseases. In our clinical and preclinical programs, we have utilized multiple isotopes including Ac-225, I-131
and Lu-177 directed at multiple targets in oncology and hematology such as CD45, CD33, CD38, HER2, among others. Our targeted radiotherapies
combine the cell-killing ability of radiation via a radioisotope payload with a targeting agent, such as a monoclonal antibody.
With our in-depth, long-term
experience in clinical development of Ac-225 based radiopharmaceuticals, we have developed an end-to-end technology solution for producing
Ac-225 that has demonstrated radiochemical and radionuclidic purity identical to current gold standard methods. This patented technology
has been used to produce Ac-225 in a cyclotron that is essentially identical to that derived from a Th-229 generator and has the potential
to be a lower-cost, commercially scalable higher-yielding approach. Using the cyclotron-produced Ac-225 technology allows for large commercial
scale production with estimated cost of goods sold including capital expenditures and operational costs for a single cyclotron facility
of between $650 and $1,000 per mCi, which is between 10 to 20 times less expensive than the price of currently available Ac-225 material.
Our extensive know-how related
to this production technology is supported by five issued patents in the U.S. and 41 patents internationally and covers:
| |
● |
End-to-end solution including processing and recycling of Radium-226 starting material |
| |
|
|
| |
● |
Production of up to 100 mCi of Ac-225 per production cycle |
| |
|
|
| |
● |
Utilization of a medium energy cyclotron |
| |
|
|
| |
● |
Expected cost 10 to 20 times lower than currently available material |
| |
|
|
| |
● |
Radiochemical purity > 99% |
| |
|
|
| |
● |
Radioisotopic purity 99.8% with no long-lived contaminants and <0.001% Ac-227 |
With our Ac-2225 based Actimab-A
program and the rapidly increasing number of Ac-225 based programs in development, we believe that we are well positioned to leverage
this technology to produce Ac-225 to address the growing clinical demand.
Manufacturing and Supply
Chain
Actinium has established significant
manufacturing and supply chain expertise due to the unique manufacturing and distribution requirements of radiotherapeutics. Due to the
short half-life of radioisotopes, the finished drug product is shipped “hot” and must be administered within days. We have
established core competencies in the process of manufacturing radiotherapeutics, coordinating with the hospital’s care team, and
delivering “just-in-time” doses. We have delivered over 500 doses for 18 clinical trials at 45 large cancer hospitals and
have never missed a dose.
Isotope supply is critical
for the manufacturing of radiotherapeutics, and we have engaged several sources for the procurement of alpha (e.g., Ac-225) and beta (e.g.,
I-131 and Lu-177) emitters. We also have multiple isotope supply agreements and qualified vendors in place to supply isotopes for commercial
production.
We have established an actively
managed end-to-end supply chain that encompasses isotope sourcing through drug administration at the point of care and as a result of
our approach, we have reliably supplied drug to all patients enrolled in our clinical trials. We believe we have a thorough understanding
and working knowledge of the intricacies required to manufacture and distribute radiotherapies. Through our clinical experience with,
we have developed a wealth of proprietary knowledge to enable coordination between us and all key stakeholders including, but not limited
to hematologists/oncologists, infusion center and in patient rooms, nuclear medicine and radiology, hot labs and radio-pharmacies, and
radiation safety committees, among others. We utilize contract development and manufacturing organizations (“CDMOs”) to manufacture
our finished drug products and have selected CDMOs based on their clinical and commercial supply capabilities including significant experience
in the international supply of radiotherapies. We have scaled deliberately for manufacturing flexibility and will continue to evaluate
additional CDMOs.
Intellectual Property
Our proprietary technology
platform is supported by IP, know-how and trade secrets that cover the generation, development, methods of use and manufacture of targeted
radiotherapies and their select components. Our IP covers various methods of use in multiple diseases, including indication, dose and
scheduling, radionuclide warhead, and therapeutic combinations.
As of November 14, 2024,
our patent portfolio is comprised of over 230 issued patents and pending patent applications worldwide, which we believe constitutes
a valuable business asset. Our IP includes 47 patent families, including key patents that relate primarily to our radiotherapeutic
candidates. Our patent portfolio includes 20 issued patents and 48 pending patent applications in the U.S., and over 165 that are
issued or pending internationally. The effective lives of the issued patents in our portfolio, or patents that may issue from the
pending applications in our portfolio, ranges from expirations between 2025 and 2045.
For our Iomab-B product candidate,
we have four issued patents in the U.S. and issued patents in Canada, Europe and Japan that relate to its composition. The basic patent
terms of these patents expire in 2036 and 2037. Related patent applications are also currently pending in the U.S. and internationally.
In addition, we own both U.S. and international pending patent applications that relate to the use of Iomab-B or Iomab-ACT with currently
issued U.S. patent protection into 2040 for the use of Iomab-ACT in connection with genetically modified hematopoietic stem cell therapy
for the treatment of non-malignant conditions.
Our patents also cover key
areas of our business such as manufacturing key components of our product candidate, Actimab-A, including producing Ac-225 in a cyclotron.
We have expertise in utilizing the alpha emitting isotope Ac-225 including clinical experience in treating approximately 150 patients
with our alpha-emitter-based therapies, “gold standard” linker technology and five issued patents in the U.S. and 41 patents
internationally related to the manufacturing of Ac-225 in a cyclotron, which we believe has the potential to produce higher quantities
of Ac-225 than currently utilized methods. In addition, we also own U.S. and international patents and pending patent applications that
relate to the manufacturing of Actimab-A and its use in the treatment of cancers.
Human and Intellectual Capital
As of November 4, 2024, we
have 37 full-time employees. Over forty percent of our employees have advanced scientific and medical degrees across our Clinical, R&D,
CMC and business and corporate development teams. In the third quarter of 2024, our overall headcount reduced by approximately twenty
percent, with a majority of these former employees being from our clinical and CMC groups. As a result of these departures, we expect
our personnel expenses to be reduced by approximately $3.7 million in 2025, which may be offset by additional hires or consultants. We
do not expect these departures to have a material impact on our operations or ability to execute our operating plan and we are actively
seeking a strategic partner for Iomab-B in the U.S. to advance the head-to-head clinical trial or other clinical development activity
for Iomab-B.
Results of Operations –
Three Months Ended September 30, 2024 Compared to Three Months Ended September 30, 2023
The following table sets forth,
for the periods indicated, data derived from our statements of operations:
| | |
For the
Three Months Ended
September 30, | |
| (in thousands) | |
2024 | | |
2023 | |
| | |
| | |
| |
| Revenue: | |
| | |
| |
| Revenue | |
$ | - | | |
$ | - | |
| Other revenue | |
| - | | |
| - | |
| Total revenue | |
| - | | |
| - | |
| | |
| | | |
| | |
| Operating expenses: | |
| | | |
| | |
| Research and development, net of reimbursements | |
| 9,774 | | |
| 11,622 | |
| General and administrative | |
| 2,827 | | |
| 2,729 | |
| Total operating expenses | |
| 12,601 | | |
| 14,351 | |
| | |
| | | |
| | |
| Other income: | |
| | | |
| | |
| Interest income – net | |
| 1,033 | | |
| 1,075 | |
| Total other income | |
| 1,033 | | |
| 1,075 | |
| | |
| | | |
| | |
| Net loss | |
$ | (11,568 | ) | |
$ | (13,276 | ) |
Revenue
We recorded no commercial
revenue for the three months ended September 30, 2024 and September 30, 2023.
Other revenue
The National Institutes of
Health awarded us a Small Business Technology Transfer cost reimbursable grant to support a clinical collaboration with Memorial Sloan
Kettering Cancer Center, or MSK, to study Iomab-ACT, our CD45-targeting Antibody Radio-Conjugate, for targeted conditioning to achieve
lymphodepletion prior to administration of a CD19-targeted CAR T-cell therapy developed at MSK. There was no other revenue recognized
for the three months ended September 30, 2024 and 2023, respectively.
As noted above, on April
7, 2022, we entered into a License Agreement with Immedica, pursuant to which Immedica licensed the exclusive product rights for commercialization
of Iomab-B in certain countries in the EUMENA region. Upon signing, we were entitled to an upfront, non-refundable payment of $35 million
from Immedica, which was received in May 2022. Under the terms of the License Agreement, we are eligible to receive certain regulatory
and commercial milestone payments and royalties on net sales of the product in certain countries that may result from the License Agreement.
We continue to retain commercialization rights in the U.S. and rest of the world.
Our contract liabilities are
recorded within Other revenue deferred – current liability or Long-term license revenue deferred in our condensed consolidated balance
sheets depending on the short-term or long-term nature of the payments to be recognized. Our contract liabilities primarily consist of
advanced payments from licensees. There was no Other revenue deferred-current liability at September 30, 2024 and December 31, 2023. Long-term
license revenue deferred was $35 million at September 30, 2024 and December 31, 2023, resulting from the receipt from Immedica; this deferred
revenue will be recognized upon the European Union’s regulatory approval of Iomab-B or provision of definitive feedback that Iomab-B
will not receive approval in the European Union.
Research and Development Expense, net of reimbursements
Research and development expenses
of $9.8 million for the three months ended September 30, 2024 decreased $1.8 million from $11.6 million for the three months ended September
30, 2023. The decrease is primarily a result of a decline in Chemistry, manufacturing and controls, or CMC, expenses of $5.5 million and
lower consulting expenses of $0.5 million, both due to lower activity in 2024 related to Iomab-B. These declines were partially offset
by increased preclinical expenses of $3.9 million.
General and administrative expense
General and administrative
expenses of $2.8 million for the three months ended September 30, 2024 increased by $0.1 million from $2.7 million for the three months
ended September 30, 2023, primarily due to higher non-cash stock compensation expense of $0.5 million, partially offset by lower compensation
expense of $0.3 million due to lower headcount.
Other income
Other income is comprised
of net interest income in both reporting periods. The amount for the three months ended September 30, 2024 and 2023 of $1.0 million in
each reporting period was virtually unchanged from the same time period in the prior year.
Net loss
Net loss of $11.6 million
for the three months ended September 30, 2024 decreased by $1.7 million from $13.3 million for the three months ended September 30, 2023
due to lower research and development expenses partially offset by higher general and administrative expenses.
Results of Operations –Nine
Months Ended September 30, 2024 Compared to Nine Months Ended September 30, 2023
The following table sets forth,
for the periods indicated, data derived from our statements of operations:
| | |
For the
Nine Months Ended
September 30, | |
| (in thousands) | |
2024 | | |
2023 | |
| | |
| | |
| |
| Revenue: | |
| | |
| |
| Revenue | |
$ | - | | |
$ | - | |
| Other revenue | |
| - | | |
| - | |
| Total revenue | |
| - | | |
| - | |
| | |
| | | |
| | |
| Operating expenses: | |
| | | |
| | |
| Research and development, net of reimbursements | |
| 25,234 | | |
| 30,552 | |
| General and administrative | |
| 9,382 | | |
| 11,025 | |
| Total operating expenses | |
| 34,616 | | |
| 41,577 | |
| | |
| | | |
| | |
| Other income: | |
| | | |
| | |
| Interest income – net | |
| 3,025 | | |
| 2,083 | |
| Total other income | |
| 3,025 | | |
| 2,083 | |
| | |
| | | |
| | |
| Net loss | |
$ | (31,591 | ) | |
$ | (39,494 | ) |
Revenue
We recorded no commercial
revenue for the nine months ended September 30, 2024 and 2023, respectively.
Other revenue
There was no other revenue
recognized for the nine months ended September 30, 2024 and 2023, respectively.
Research and Development Expense, net of reimbursements
Research and development expenses
of $25.2 million for the nine months ended September 30, 2024 decreased $5.4 million from $30.6 million for the nine months ended September
30, 2023. CMC expenses declined $9.7 million and consulting expenses declined $1.7 million due to lower CMC activity related to Iomab-B.
These declines were partially offset by increased preclinical expenses of $4.9 million, and as a result of higher headcount, increased
compensation expense of $0.8 million and non-cash stock compensation expense of $0.2 million.
General and administrative expense
General and administrative
expenses of $9.4 million for the nine months ended September 30, 2024 decreased by $1.6 million from $11.0 million for the nine months
ended September 30, 2023. Lower expenses were primarily the result of lower consulting fees and legal fees of $1.7 million, lower compensation
expense of $0.5 million due to lower headcount, and lower office expenses of $0.5 million, partially offset by higher non-cash stock compensation
expense of $1.0 million.
Other income
Other income is comprised
of net interest income in both reporting periods. The amount for the nine months ended September 30, 2024 of $3.0 million increased $0.9
million from $2.1 million for the nine months ended September 30, 2023 due to higher average interest rates.
Net loss
Net loss of $31.6 million
for the nine months ended September 30, 2024 decreased by $7.9 million from $39.5 million for the nine months ended September 30, 2023
due to lower research and development expenses, lower general and administrative expenses and a higher level of other income.
Liquidity and Capital Resources
Historically, we have financed
our operations primarily through sales of shares of our stock. The following table sets forth selected cash flow information for the periods
indicated:
| | |
For the
Nine Months Ended
September 30, | |
| (in thousands) | |
2024 | | |
2023 | |
| | |
| | |
| |
| Cash used in operating activities | |
$ | (27,321 | ) | |
$ | (39,845 | ) |
| Cash used in investing activities | |
| (11 | ) | |
| (153 | ) |
| Cash provided by financing activities | |
| 29,324 | | |
| 13,678 | |
| | |
| | | |
| | |
| Net change in cash, cash equivalents and restricted cash | |
$ | 1,992 | | |
$ | (26,320 | ) |
Net cash used in operating
activities for the nine months ended September 30, 2024 of $27.3 million decreased by $13.5 million from $39.8 million in the prior-year
period, primarily as a result of a lower net loss of $7.9 million, and compared to the prior-year period, a reduction in prepaid expenses
of $1.7 million and an increase in accounts payable and accrued expenses of $1.2 million.
Net cash used in investing
activities was $11 thousand and $153 thousand for the nine months ended September 30, 2024 and 2023, respectively, due to the purchase
of equipment.
Net cash provided by financing
activities was $29.3 million and $13.7 million for the nine months ended September 30, 2024 and 2023, respectively, primarily from the
sale of common stock.
In August 2020, we entered
into the Capital on Demand™ Sales Agreement with JonesTrading Institutional Services LLC, or JonesTrading, pursuant to which we
are able to sell, from time to time, through or to JonesTrading, up to an aggregate of $200 million of our common stock. On June 28, 2022,
we entered into an Amendment and Restated Capital on Demand™ Sales Agreement, or the Amended Sales Agreement, with JonesTrading
and B. Riley Securities, Inc. The Amended Sales Agreement modifies the original Capital on Demand™ Sales Agreement to include B.
Riley as an additional sales agent thereunder. Shares of common stock are offered pursuant to a shelf registration statement on Form S-3
(File No. 333-273911), which was declared effective February 5, 2024, including a base prospectus covering the offering, issuance and
sale of up to $500 million of common stock, preferred stock, warrants, units and/or subscription rights; and a sales agreement prospectus
covering the offering, issuance and sale of up to a maximum aggregate offering price of $200 million of common stock that may be issued
and sold under the Amended Sales Agreement. For the nine months ended September 30, 2024, we sold 3.5 million shares of common stock,
resulting in gross proceeds of $29.9 million and net proceeds of $29.3 million. For the nine months ended September 30, 2023, we sold
3.0 million shares of common stock, resulting in gross proceeds of $18.9 million and net proceeds of $18.3 million.
As of the date of filing this
report, we expect that our existing resources will be sufficient to fund our planned operations for more than 12 months following the
date of this report.
Critical Accounting Estimates
Our management’s discussion
and analysis of financial condition and results of operations is based on our consolidated financial statements, which have been prepared
in accordance with accounting principles generally accepted in the United States, (“GAAP”). The preparation of these financial
statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses and the disclosure
of contingent assets and liabilities in our consolidated financial statements during the reporting periods. These items are monitored
and analyzed by us for changes in facts and circumstances, and material changes in these estimates could occur in the future. We base
our estimates on historical experience, known trends and events, and on various other factors that we believe are reasonable under the
circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not
readily apparent from other sources. Changes in estimates are reflected in reported results for the period in which they become known.
Actual results may differ materially from these estimates under different assumptions or conditions. The Company does not have any critical
accounting estimates.
Recently Issued Accounting Pronouncements
In December 2023, FASB issued
ASU 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures, to enhance the transparency and decision usefulness
of income tax disclosures. The amendments in ASU 2023-09 provide improvements primarily related to the rate reconciliation and income
taxes paid information included in income tax disclosures. We would be required to disclose additional information regarding reconciling
items equal to or greater than five percent of the amount computed by multiplying pretax income (loss) by the applicable statutory tax
rate. Similarly, we would be required to disclose income taxes paid (net of refunds received) equal to or greater than five percent of
total income taxes paid (net of refunds received). The amendments in ASU 2023-09 are
effective January 1, 2025, including interim periods. Early adoption is permitted for annual financial statements that have not yet been
issued or made available for issuance. We will evaluate the impact of ASU 2023-09 on our financial statements.
In November 2023, FASB issued
ASU 2023-07, Segment Reporting (Topic 280), Improvements to Reportable Segment Disclosures, which provides improvements to reportable
segment disclosure requirements, primarily through enhanced disclosures around segment expenses. ASU 2023-07 requires us to disclose significant
segment expenses that are regularly provided to the chief operating decision maker, or CODM, and included within each reported measure
of segment profit or loss. ASU 2023-07 also requires that we disclose an amount for other segment items by reportable segment, a description
of their composition and provide all annual disclosures about a reportable segment’s profit or loss and assets pursuant to Topic
280 during interim periods. We must also disclose the CODM’s title and position, as well as certain information around the measures
used by the CODM and an explanation of how the CODM uses the reported measures in assessing segment performance and deciding how to allocate
resources. For public entities with a single reportable segment, the entity must provide all the disclosures required pursuant to ASU
2023-07 and all existing segment disclosures under Topic 280. The amendments of ASU 2023-07 are
effective for us for annual periods beginning January 1, 2024, and effective for interim periods beginning January 1, 2025. We will adopt
this standard effective January 1, 2024, and report on it in our Annual Report on Form 10-K for the year ended December 31, 2024. We will
update all required disclosures pursuant to ASU 2023-07 at that time.
In October 2021, FASB issued
ASU 2021-08, Business Combinations (Topic 805), Account for Contract Assets and Contract Liabilities from Contracts with Customers,
which provides guidance on accounting for contract assets and contract liabilities acquired in a business combination in accordance
with ASC 606. To achieve this, an acquirer may assess how the acquiree applied ASC 606 to determine what to record for the acquired revenue
contracts. Generally, this should result in an acquirer recognizing and measuring the acquired contract assets and contract liabilities
consistent with how they were recognized and measured in the acquiree’s financial statements. The amendments of ASU 2021-08 were
effective January 1, 2023, including interim periods. We will evaluate the impact of ASU 2021-08 on any future business combinations we
may enter in the future.
ITEM 3. QUANTITATIVE AND QUALITATIVE DISCLOSURES
ABOUT MARKET RISK.
We are not currently exposed
to significant market risk related to changes in interest rates. As of September 30, 2024, our cash equivalents consisted primarily of
short-term money market funds. Our primary exposure to market risk is interest rate sensitivity, which is affected by changes in the general
level of U.S. interest rates. Due to the short-term nature of the cash equivalents in our portfolio and the low risk profile of our cash
equivalents, an immediate 10% change in interest rates would not have a material effect on the fair market value of our financial position
or results of operations.
We are not currently exposed
to significant market risk related to changes in foreign currency exchange rates. Our operations may be subject to fluctuations in foreign
currency exchange rates in the future.
Inflation generally affects
us by increasing our cost of labor and clinical trial costs. We do not believe that inflation had a material effect on our business, financial
condition or results of operations during the nine months ended September 30, 2024 and 2023.
ITEM 4. CONTROLS AND PROCEDURES.
Evaluation of Disclosure
Controls and Procedures. Under the supervision and with the participation of our management, including our principal executive
officer and principal financial and accounting officer, we conducted an evaluation of the effectiveness, as of September 30, 2024, of
our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended,
or the Exchange Act. Based upon such evaluation, our principal executive officer and principal financial and accounting officer have concluded
that, as of September 30, 2024, our disclosure controls and procedures were effective to provide reasonable assurance that the information
we are required to disclose in our filings with the Securities and Exchange Commission, or SEC, under the Exchange Act (i) is recorded,
processed, summarized and reported within the time periods specified in the SEC’s rules and forms and (ii) accumulated and communicated
to our management, including our principal executive officer and principal financial and accounting officer, as appropriate to allow timely
decisions regarding required disclosure.
Changes in Internal Control
over Financial Reporting. There were no changes in our internal controls over financial reporting
during the period covered by this Quarterly Report of Form 10-Q that has materially affected, or are reasonably likely to materially affect,
our internal control over financial reporting.
PART II – OTHER INFORMATION
ITEM 1. LEGAL PROCEEDINGS
None.
ITEM 1A. RISK FACTORS
In analyzing our company,
you should consider carefully the following risk factors, together with all of the other information included in this Quarterly Report
on Form 10-Q. Factors that could cause or contribute to differences in our actual results include those discussed in the following
subsection, as well as those discussed above in “Management’s Discussion and Analysis of Financial Condition and Results of
Operations” and in our Annual Report filed on Form 10-K for the year ended December 31, 2023. Each of the following risk factors,
either alone or taken together, could adversely affect our business, operating results and financial condition, as well as adversely affect
the value of an investment in our company. The risks and uncertainties described below are not the only ones we face. Additional risks
not currently known to us or other factors not perceived by us to present significant risks to our business at this time also may impair
our business operations.
Summary of Risk Factors
We are providing the following
summary of the risk factors contained in this Quarterly Report on Form 10-Q to enhance the readability and accessibility of our risk factor
disclosures. We encourage you to carefully review the full risk factors contained in this Quarterly Report on Form 10-Q in their entirety
for additional information regarding the material factors that make an investment in our securities speculative or risky. These risks
and uncertainties include, but are not limited to, the following:
| |
● |
We are a clinical-stage company and have generated no revenue from commercial sales to date; |
| |
|
|
| |
● |
We have incurred net losses in every year since our inception and anticipate that we will continue to incur net losses in the future; |
| |
|
|
| |
● |
If we fail to obtain additional financing, we will be unable to continue or complete our product development or product commercialization and you will likely lose your entire investment; |
| |
|
|
| |
● |
We are highly dependent on the clinical, regulatory and commercial
success of Iomab-B, Actimab-A, Iomab-ACT and other pipeline candidates which we may never achieve; |
| |
|
|
| |
● |
We continuously evaluate our business strategy and may modify our strategy
as necessary to respond to developments in our business and other factors, and any such modification, if not successful, could have a
material adverse effect on our business, financial condition, and results of operations; |
| |
|
|
| |
● |
Our business could be adversely affected by the effects of future health epidemics; |
| |
|
|
| |
● |
Our business is subject to cybersecurity risk; |
| |
|
|
| |
● |
We have not demonstrated that any of our products are safe and effective for any indication and will continue to expend substantial time and resources on clinical development before any of our current or future product candidates will be eligible for FDA approval, if ever; |
| |
|
|
| |
● |
Our clinical trials may fail to demonstrate adequately the efficacy and safety of our product candidates, which would prevent or delay regulatory approval and commercialization; |
| |
|
|
| |
● |
Preliminary, Interim, and “top-line” data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.; |
| |
● |
Healthcare legislative reform measures intended to increase pressure to reduce prices of pharmaceutical products paid for by Medicare or, otherwise, affect the federal regulation of the U.S. healthcare system could have a material adverse effect our business, future revenue, if any, and results of operations; |
| |
● |
We may rely on third parties to conduct certain aspects of our clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines or comply with regulatory requirements, we may not be able to obtain regulatory approval for or commercialize our product candidates; |
| |
|
|
| |
● |
We currently depend on single third-party manufacturers to produce our pre-clinical and clinical trial drug supplies. Any disruption in the operations of our current third-party manufacturers, or other third-party manufacturers we may engage in the future, could adversely affect our business and results of operations; |
| |
● |
Our product candidates may cause undesirable side effects or have other properties that could halt their clinical development, prevent their regulatory approval, limit their commercial potential, or result in significant negative consequences; |
| |
|
|
| |
● |
Our patent position is highly uncertain and involves complex legal and factual questions. |
| |
|
|
| |
● |
The use of hazardous materials, including radioactive and biological materials, in our research and development efforts imposes certain compliance costs on us and may subject us to liability for claims arising from the use or misuse of these materials; |
| |
|
|
| |
● |
We are highly dependent on our key personnel, and the demand for talent in the biotechnology industry is highly competitive; if we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement or execute our business strategy; |
| |
|
|
| |
● |
Certain provisions of our Certificate of Incorporation and Bylaws and Delaware law make it more difficult for a third party to acquire us and make a takeover more difficult to complete, even if such a transaction were in our stockholders’ interest; and |
| |
|
|
| |
● |
Our ability to utilize our net operating loss carryforwards and certain other tax attributes may be limited. |
Risks Related to Our Business
We are a clinical-stage company and have
generated no revenue from commercial sales to date.
We are a clinical-stage biopharmaceutical
company with a limited operating history. We have no products approved for commercial sale and have not generated any revenue from product
sales to date. We will encounter risks and difficulties frequently experienced by early-stage companies in rapidly evolving fields. If
we do not address these risks successfully, our business will suffer.
We have incurred net losses in every year
since our inception and anticipate that we will continue to incur net losses in the future.
We are not profitable and
have incurred losses in each period since our inception. As of September 30, 2024 and December 31, 2023, we had an accumulated deficit
of $369.2 million and $337.6 million, respectively. We reported a net loss of $31.6 million and $39.5 million for the nine months ended
September 30, 2024 and 2023, respectively. We expect to continue to operate at a net loss as we continue our research and development
efforts, continue to conduct clinical trials and develop manufacturing, sales, marketing and distribution capabilities. There can be no
assurance that the products under development by us will be approved for sale in the United States or elsewhere. Furthermore, there can
be no assurance that if such products are approved, they will be successfully commercialized, which would have an adverse effect on our
business prospects, financial condition and results of operation.
If we fail to obtain additional financing,
we will be unable to continue or complete our product development and you will likely lose your entire investment.
As of the date of filing this
report, we expect that our existing resources will be sufficient to fund our planned operations for more than 12 months following the
date of this report.
Our business or operations
may change in a manner that would consume available funds more rapidly than anticipated and substantial additional funding may be required
to maintain operations, fund expansion, develop new or enhanced products, acquire complementary products, business or technologies or
otherwise respond to competitive pressures and opportunities, such as a change in the regulatory environment or a change in preferred
cancer treatment modalities. However, we may not be able to secure funding when we need it or on favorable terms or indeed on any terms.
In addition, from time to time, we may not be able to secure enough capital in a timely enough manner which may cause the generation of
a going-concern opinion from our auditors which can and may impair our stock market valuation and also our ability to finance on favorable
terms or indeed on any terms.
To raise additional capital,
we may in the future offer additional shares of our common stock or other securities convertible into or exchangeable for our common stock.
We cannot assure you that we will be able to sell shares or other securities in any other offering at a price per share that is equal
to or greater than the price per share paid by investors, and investors purchasing shares or other securities in the future could have
rights superior to existing stockholders.
If we cannot raise adequate
funds to satisfy our capital requirements, we will have to delay, scale back or eliminate our research and development activities, clinical
studies, or future operations. We may also be required to obtain funds through arrangements with collaborators, which arrangements may
require us to relinquish rights to certain technologies or products that we otherwise would not consider relinquishing, including rights
to future product candidates or certain major geographic markets. We may further have to license our technology to others. This could
result in sharing revenues which we might otherwise have retained for ourselves. Any of these actions may harm our business, financial
condition, and results of operations.
The amount of funding we will
need depends on many factors, including the progress, timing and scope of our product development programs; the progress, timing and scope
of our preclinical studies and clinical trials; the time and cost necessary to obtain regulatory approvals; the time and cost necessary
to further develop manufacturing processes and arrange for contract manufacturing; our ability to enter into and maintain collaborative,
licensing and other commercial relationships; and our partners’ commitment of time and resources to the development and commercialization
of our products.
We have limited access to the capital markets
and even if we can raise additional funding, we may be required to do so on unfavorable terms.
We have limited access to
the capital markets to raise funds. The capital markets have been unpredictable in the recent past for development stage radiopharmaceutical
and other biotechnology companies and unprofitable companies such as ours. In addition, it is generally difficult for development-stage
companies to raise capital under current market conditions. The amount of capital that a company such as ours is able to raise often depends
on variables that are beyond our control. As a result, we may not be able to secure financing on terms attractive to us, or at all. If
we are able to consummate a financing arrangement, the amount raised may not be sufficient to meet our future needs. If adequate funds
are not available on acceptable terms, or at all, our business, including our technology licenses, results of operations, financial condition
and our continued viability will be materially adversely affected.
We are highly dependent on the clinical,
regulatory and commercial success of Iomab-B, Actimab-A, Iomab-ACT and other pipeline candidates which we may never achieve
None of the drug candidates
we are developing, or have developed, have received regulatory approval. Based on the current status of our pipeline candidates, it will
likely take several years or additional clinical studies before we can seek approval for any drug candidate.
As for Iomab-B in
particular, as previously disclosed, we completed the pivotal Phase 3 SIERRA trial (Study of Iomab-B in Elderly Relapsed or
Refractory AML) and presented the trial results in February 2023, which were expected to support a BLA filing. The SIERRA trial met
the primary endpoint of dCR with statistical significance (p-value<0.0001) but did not meet the secondary endpoint in achieving a
statistically significant improvement in overall survival in the intent to treat population. On August 5, 2024, we announced that
the FDA determined that the SIERRA trial alone is not adequate to support a BLA filing and is requiring an additional randomized
head-to-head clinical trial to demonstrate an overall survival benefit in an intent to treat population. Further, the FDA now
requires an additional dose optimization trial to calculate the dose of Iomab-B based on absorbed dose by the bone marrow, rather
than the maximum tolerable dose of 24 Gy of radiation to the liver as was done in the SIERRA trial based on several interactions
with the FDA prior to the start of the SIERRA trial. Based on this revised approach now required by the FDA, the safety and efficacy
data generated from all Iomab-B studies, including the SIERRA trial, are inadequate to seek regulatory approval for Iomab-B, as
dosing based on maximum tolerable dose of 24 Gy to the liver will lead to variable doses to the bone marrow (the target organ),
result in underdosing or overdosing of patients and translate to a global patient safety risk. We are seeking a strategic partner
for the U.S. in order to conduct the additional studies required by the FDA; however, we may not be successful in our efforts to
find such a partner, or the trials and studies may not be successful. Further, there are no assurances that we can satisfy all of
the FDA’s requests, and there could be additional regulatory hurdles that may result in either non-acceptance or non-approval
of a future BLA filing. The U.S. commercial opportunity for Iomab-B may thus never be realized.
As previously disclosed and noted
above, Actinium has licensed to Immedica the exclusive product rights for commercialization of Iomab-B in the EUMENA region. We are evaluating
the impact of the FDA’s 2024 determination of the SIERRA trial results in the context of global regulatory submissions for Iomab-B
and at this time there can be no assurance of filings for regulatory approval, obtaining regulatory approvals, or successful commercialization
of Iomab-B including on a global basis.
Our Actimab-A drug candidate
was studied in a Phase 2 clinical trial as a monotherapy and we are now studying it in combination with other therapies. We have aligned
with the FDA on an operationally seamless Phase 2/3 trial that is intended to support a BLA filing. There can be no assurance that the
Phase 2 portion of the trial will be successful and support advancing to the Phase 3 portion of the trial. In addition, our Iomab-ACT
drug candidate has only been studied in a limited number of human subjects in a Phase 1 trial with a novel CAR-T therapy. While the initial
results from this trial were encouraging, there can be no assurance that future results with Iomab-ACT from the commercial CAR-T trial
at UTSW or sickle cell conditioning trial at Columbia will be positive.
We may be unable to establish sales, marketing
and commercial supply capabilities.
We do not currently have,
nor have we ever had, commercial sales and marketing capabilities. If any of our product candidates ultimately become approved and we
do not secure a commercial partner, we would have to build and establish these capabilities in order to commercialize our approved product
candidates. The process of establishing commercial capabilities will be expensive and time consuming. Even if we are successful in building
sales and marketing capabilities, we may not be successful in commercializing any of our product candidates. Any delays in commercialization
or failure to successfully commercialize any product candidate may have material adverse impacts on our business and ability to continue
operations.
Our business could be adversely affected
by the effects of future health epidemics.
Our business could be adversely
impacted by the effects of future pandemics, epidemics or infectious disease outbreaks. The full impact of such an event cannot be predicted
at this time, and could depend on numerous factors, including vaccination rates among the population and the response by governmental
bodies and regulators. Given the ongoing and dynamic nature of the circumstances, it is difficult to predict the impact of a future pandemic
on our business.
A future pandemic could adversely
affect our clinical trial operations, including our ability to conduct the trials on the expected timelines and recruit and retain patients
and principal investigators and site staff who, as healthcare providers, may have heightened exposure to a future pandemic if their geography
is impacted by the pandemic. Further, future pandemics could result in delays in our clinical trials due to prioritization of hospital
resources toward the pandemic, restrictions in travel, potential unwillingness of patients to enroll in trials, or the inability of patients
to comply with clinical trial protocols if quarantines or travel restrictions are implemented that impede patient movement or interrupt
healthcare services. In addition, we rely on independent clinical investigators, contract research organizations and other third-party
service providers to assist us in managing, monitoring and otherwise carrying out our preclinical studies and clinical trials, and a future
pandemic may affect their ability to devote sufficient time and resources to our programs or to travel to sites to perform work for us,
which may result in delays or hinder our ability to collect data from our clinical trials.
Additionally, a future pandemic
may result in delays in receiving approvals from local and foreign regulatory authorities, delays in necessary interactions with IRB’s
or Institutional Review Boards, local and foreign regulators, ethics committees and other important agencies and contractors due to limitations
in employee resources or forced furlough of government employees.
Our business is subject to cybersecurity
risks.
Our operations are increasingly
dependent on information technologies and services. Threats to information technology systems associated with cybersecurity risks and
cyber incidents or attacks continue to grow, and include, among other things, storms and natural disasters, terrorist attacks, utility
outages, theft, viruses, phishing, malware, design defects, human error, and complications encountered as existing systems are maintained,
repaired, replaced, or upgraded. Risks associated with these threats include, among other things:
| |
● |
theft or misappropriation of funds; |
| |
|
|
| |
● |
loss, corruption, or misappropriation of intellectual property, or other proprietary, confidential or personally identifiable information (including supplier, clinical data or employee data); |
| |
|
|
| |
● |
disruption or impairment of our and our business operations and safety procedures; |
| |
● |
damage to our reputation with our potential partners, patients and the market; |
| |
|
|
| |
● |
exposure to litigation; |
| |
|
|
| |
● |
increased costs to prevent, respond to or mitigate cybersecurity events. |
Although we utilize various
procedures and controls to mitigate our exposure to such risk, cybersecurity attacks and other cyber events are evolving and unpredictable.
Moreover, we have no control over the information technology systems of third parties conducting our clinical trials, our suppliers, and
others with which our systems may connect and communicate. As a result, the occurrence of a cyber incident could go unnoticed for a period
time.
We have cybersecurity insurance
coverage in the event we become subject to various cybersecurity attacks, however, we cannot ensure that it will be sufficient to cover
any particular losses we may experience as a result of such cyberattacks. Any cyber incident could have a material adverse effect on our
business, financial condition and results of operations.
Risks Related to Regulation
The FDA, EMA or comparable foreign regulatory
authorities may disagree with our regulatory plans and we may fail to obtain regulatory approval of our product candidates.
Our products are subject to
rigorous regulation by the FDA, EMA and numerous other federal, state and foreign governmental authorities. The process of seeking regulatory
approval to market an antibody radiation-conjugate product is expensive and time-consuming, and, notwithstanding the effort and expense
incurred, approval is never guaranteed. If we are not successful in obtaining timely approval of our products from the regulators, we
may never be able to generate significant revenue and may be forced to cease operations. In particular, the FDA permits commercial distribution
of a new antibody radiation-conjugate product only after a BLA for the product has received FDA approval. The BLA process is costly, lengthy
and inherently uncertain. Any BLA filed by us will have to be supported by extensive data, including, but not limited to, technical, preclinical,
clinical trial, chemistry, manufacturing and controls and labeling data, to demonstrate to the FDA’s satisfaction the safety and
efficacy of the product for its intended use. The lengthy approval process as well as the unpredictability of future clinical trial results
may result in our failing to obtain regulatory approval to market our product candidates, which would significantly harm our business,
results of operations and prospects. In addition, even if we were to obtain approval, regulatory authorities may approve any of our product
candidates for fewer or more limited indications than we request, may not obtain the price we intend to charge for our products, may grant
approval contingent on the performance of costly post-marketing clinical trials, or may approve a product candidate with a label that
does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. Any of the
foregoing scenarios could materially harm the commercial prospects for our product candidates.
For instance, as for Iomab-B,
despite the Phase SIERRA 3 trial meeting the primary endpoint of durable Complete Remission (dCR) with statistical significance (p-value<0.0001),
the FDA has determined that demonstrating an overall survival benefit in a randomized head-to-head trial is required for a BLA filing.
In addition, the FDA now requires that an additional dose optimization trial demonstrating safety and efficacy be completed to calculate
the dose of Iomab-B based on absorbed dose by the bone marrow, rather than the maximum tolerable dose of 24 Gy of radiation to the liver
as was done in the SIERRA trial based on several interactions we had with the FDA before starting the SIERRA trial. The head-to-head Phase
3 trial will evaluate allogeneic bone marrow transplant (BMT) using Iomab-B plus a reduced intensity conditioning regimen of fludarabine
and total body irradiation (Flu/TBI) versus allogeneic BMT using reduced intensity conditioning comprised of cyclophosphamide plus Flu/TBI.
This is different from the SIERRA trial, which allowed physician’s choice of salvage therapies and heterogenous conditioning regimens
in the control arm. However, there are no assurances that the additional trials will be successful or that we can satisfy all of the FDA’s
requests. There could be additional regulatory hurdles that may result in either non-acceptance or non-approval of a future BLA filing.
As previously disclosed and noted
above, Actinium has licensed to Immedica the exclusive product rights for commercialization of Iomab-B in the EUMENA region. We are evaluating
the impact of the FDA’s 2024 determination of the SIERRA trials results on global regulatory submissions for Iomab-B and at this
time, there can be no assurance of global filings for regulatory approval, obtaining regulatory approvals, or successful commercialization
of Iomab-B on a global basis.
We are also evaluating Iomab-ACT,
which uses a lower dose I-131 for conditioning prior to cellular therapies such as CAR-T and gene therapies. We are currently studying
Iomab-ACT in three clinical trials including two investigator sponsored studies.
Our CD33 Alpha program studying
Actimab-A (lintuzumab-Ac-225) product candidate has also been studied in several Phase 1 and 2 trials under our sponsorship and investigator-initiated
trials in patients with r/r AML and plan to continue to study Actimab-A in clinical trials. Actimab-A is also being developed under a
cooperative research and development agreement (CRADA) with the National Cancer Institute (NCI) and we expect clinical trials to be initiated
that will study Actimab-A as a single agent or in combination with other therapies. Product candidates utilizing the lintuzumab antibody
would require BLA approval before they can be marketed in the United States. We are in the early stages of evaluating other product candidates
consisting of conjugates of Ac-225 with human or humanized antibodies for pre-clinical and clinical development in other types of cancer.
The FDA may not approve these products for the indications that are necessary or desirable for successful commercialization. The FDA may
fail to approve any BLA we submit for new product candidates or for new intended uses or indications for approved products or future product
candidates. Failure to obtain FDA approval for our products in the proposed indications would have a material adverse effect on our business
prospects, financial condition and results of operations.
The approval process in the
United States and in other countries could result in unexpected and significant costs for us and consume management’s time and other
resources. The FDA, EMA and other foreign regulatory agencies could ask us to supplement our submissions, collect non-clinical data, conduct
additional clinical trials or engage in other time-consuming actions, or it could simply deny our applications. In addition, even if we
obtain approval to market our products in the United States or in other countries, the approval could be revoked, or other restrictions
imposed if post-market data demonstrates safety issues or lack of effectiveness. We cannot predict with certainty how, or when, the FDA, EMA or other regulatory authorities will act. If we are unable to obtain the necessary regulatory approvals, our financial condition
and cash flow may be materially adversely affected, and our ability to grow domestically and internationally may be limited. Additionally,
even if we obtain approval, regulatory authorities may approve any of our product candidates for fewer or more limited indications that
we request. The Company’s products may not be approved for the specific indications that are most necessary or desirable for successful
commercialization or profitability.
We have not demonstrated that any of our
products are safe and effective for any indication and will continue to expend substantial time and resources on clinical development
before any of our current or future product candidates will be eligible for FDA approval, if ever.
We expect that a substantial
portion of our efforts and expenditures over the next few years will be devoted to development of our existing and contemplated biological
product candidates. Accordingly, our business currently depends heavily on the successful development, FDA approval, and commercialization
of such candidates, which may never receive FDA approval or be successfully commercialized even if FDA approval is received. The research,
testing, manufacturing, labeling, approval, sale, marketing, and distribution of our biological product candidates are, and will remain,
subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries, as applicable. We
are currently not permitted to market any of our current or future product candidates in the United States until we receive FDA approval
(of each) via the BLA process. To date, we have three product candidates in clinical development and have not-yet submitted a BLA for
any of our candidates and, for many such candidates, do not expect to be in a position to do so for the foreseeable future, as there are
numerous developmental steps that must be completed before we can prepare and submit a BLA.
In the United States, the
FDA regulates pharmaceutical and biological product candidates under the FDCA and the Public Health Service Act (“PHSA”),
as well as their respective implementing regulations. Such products and product candidates are also subject to other federal, state, and
local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal,
state, local, and foreign statutes and regulations requires the expenditure of substantial time and financial resources. The process required
by the FDA before a drug or biological product may be marketed in the United States generally involves the following:
| |
● |
completion of preclinical laboratory tests and animal studies in accordance with FDA’s good laboratory practices (“GLPs”) and applicable requirements for the humane use of laboratory animals or other applicable regulations; |
| |
● |
submission to the FDA of an Investigational New Drug (“IND”), which must become effective before human clinical trials in the United States may begin; |
| |
● |
performance of adequate and well-controlled human clinical trials in
accordance with FDA’s IND regulations, good clinical practices (“GCPs”), and any additional requirements for the protection
of human research subjects and their health information, to establish the safety and efficacy of the proposed biological product for its
intended use; |
| |
|
|
| |
● |
submission to the FDA of a BLA for marketing approval that meets applicable requirements to ensure the continued safety, purity, and potency of the product that is the subject of the BLA based on results of preclinical testing and clinical trials; |
| |
|
|
| |
● |
satisfactory completion of an FDA inspection of the manufacturing facility
or facilities where the biological product is produced, to assess compliance with current good manufacturing practices (“cGMPs”)
and assure that the facilities, methods and controls are adequate to preserve the biological product’s identity, strength, quality
and purity; |
| |
● |
potential FDA audit of the nonclinical study and clinical trial sites that generated the data in support of the BLA; and |
| |
|
|
| |
● |
FDA review and approval, or denial, of the BLA. |
Before testing any biological
product candidate in humans, the product candidate enters the preclinical testing stage. Preclinical tests include laboratory evaluations
of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the product candidate.
The conduct of the preclinical tests must comply with federal regulations and requirements including GLPs. The clinical trial sponsor
must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data
or literature and a proposed clinical protocol, to the FDA as part of the IND. Some preclinical testing may continue even after the IND
is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions regarding
the proposed clinical trials and places the trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and
the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a biological
product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical
hold, trials may not recommence without FDA authorization and then only under terms authorized by the FDA. Accordingly, we cannot be sure
that submission of an IND will result in the FDA allowing clinical trials to begin or that, for those that have already commenced under
an active IND, that issues will not arise that suspend or terminate such trials.
Clinical trials involve the
administration of the biological product candidate to healthy volunteers or patients under the supervision of qualified investigators,
generally physicians not employed by or under the trial sponsor’s control. Clinical trials are conducted under protocols detailing,
among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters
to be used to monitor subject safety, including stopping rules that assure a clinical trial will be stopped if certain adverse events
should occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND. Clinical trials must be
conducted and monitored in accordance with the FDA’s regulations composing the GCP requirements, including the requirement that
all research subjects provide informed consent. Further, each clinical trial must be reviewed and approved by an independent institutional
review board, or IRB, at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting
the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical
trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed
consent that must be signed by each clinical trial subject or his or her legal representative and must monitor the clinical trial until
completed. Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| |
● |
Phase 1. The biological product is initially introduced into healthy human subjects and tested for safety. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in subjects. |
| |
● |
Phase 2. The biological product is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule. |
| |
|
|
| |
● |
Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency, and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk to benefit ratio of the product and provide an adequate basis for product labeling. |
Post-approval clinical trials,
sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These clinical trials are used to
gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up.
After the completion of clinical
trials of a biological product, FDA approval of a BLA must be obtained before commercial marketing of the biological product. The BLA
must include results of product development, laboratory and animal studies, human trials, information on the manufacture and composition
of the product, proposed labeling and other relevant information. The FDA may grant deferrals for submission of data, or full or partial
waivers. The testing and approval processes require substantial time and effort and there can be no assurance that the FDA will accept
the BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all. Before approving a BLA, the FDA
will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing
processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required
specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure that the clinical
trials were conducted in compliance with IND trial requirements and GCP requirements. To assure cGMP and GCP compliance, an applicant
must incur significant expenditure of time, money and effort in the areas of training, record keeping, production, and quality control.
Notwithstanding the submission
of relevant data and information, the FDA may ultimately decide that the BLA does not satisfy its regulatory criteria for approval and
deny approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret
the same data. We cannot predict with any certainty if or when we might submit a BLA for regulatory approval for our product candidates
or whether any such BLA will be approved by the FDA. Human clinical trials are very expensive and difficult to design and implement, in
part because they are subject to rigorous regulatory requirements. For example, the FDA may not agree with our proposed endpoints for
any clinical trial we propose, which may delay the commencement of our clinical trials. The clinical trial process is also lengthy and
requires substantial time and effort.
We expect that the clinical
trials we need to conduct to be in a position to submit BLAs for our product candidates currently in-development will take, at least,
several years to complete. Moreover, failure can occur at any stage of the trials, and we could encounter problems that cause us to abandon
or repeat clinical trials. Also, the results of early preclinical and clinical testing may not be predictive of the results of subsequent
clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials
due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier studies, and preclinical and clinical
data are often susceptible to multiple interpretations and analyses. Many companies that have believed their product candidates performed
satisfactorily in preclinical studies and clinical trials have, nonetheless, failed to obtain marketing approval of their products. Success
in preclinical testing and early clinical trials does not ensure that later clinical trials, which involve many more subjects, and the
results of later clinical trials may not replicate the results of prior clinical trials and preclinical testing. Any failure or substantial
delay in our product development plans may have a material adverse effect on our business.
We may encounter substantial delays in our
clinical trials or may not be able to conduct our trials on the timelines we expect.
We cannot predict whether
we will encounter problems with any of our ongoing or planned clinical trials that will cause us or regulatory authorities to delay, suspend,
or discontinue clinical trials or to delay the analysis of data from ongoing clinical trials. Any of the following could delay or disrupt
the clinical development of our product candidates and potentially cause our product candidates to fail to receive regulatory approval:
| |
● |
conditions imposed on us by the FDA or comparable foreign authorities regarding the scope or design of our clinical trials; |
| |
● |
delays in receiving, or the inability to obtain, required approvals from IRBs or other reviewing entities at clinical sites selected for participation in our clinical trials; |
| |
● |
delays in enrolling patients into clinical trials; |
| |
|
|
| |
● |
a lower than anticipated retention rate of patients in clinical trials; |
| |
|
|
| |
● |
the need to repeat or discontinue clinical trials as a result of inconclusive or negative results or unforeseen complications in testing or because the results of later trials may not confirm positive results from earlier preclinical studies or clinical trials; |
| |
|
|
| |
● |
inadequate supply, delays in distribution, deficient quality of, or inability to purchase or manufacture drug product, comparator drugs or other materials necessary to conduct our clinical trials; |
| |
● |
unfavorable FDA or other foreign regulatory inspection and review of a clinical trial site or records of any clinical or preclinical investigation; |
| |
|
|
| |
● |
serious and unexpected drug-related side effects experienced by participants in our clinical trials, which may occur even if they were not observed in earlier trials or only observed in a limited number of participants; |
| |
|
|
| |
● |
a finding that the trial participants are being exposed to unacceptable health risks; |
| |
|
|
| |
● |
the placement by the FDA or a foreign regulatory authority of a clinical hold on a trial; or |
| |
|
|
| |
● |
delays in obtaining regulatory agency authorization for the conduct of our clinical trials. |
We may suspend, or the FDA
or other applicable regulatory authorities may require us to suspend, clinical trials of a product candidate at any time if we or they
believe the patients participating in such clinical trials, or in independent third-party clinical trials for drugs based on similar technologies,
are being exposed to unacceptable health risks including but not limited to unacceptable or suboptimal factors related to toxicity, clinical
efficacy, imbalances in safety and efficacy profiles or for other reasons.
Further, individuals involved
with our clinical trials may serve as consultants to us from time to time and receive stock options or cash compensation in connection
with such services. If these relationships and any related compensation to the clinical investigator carrying out the study result in
perceived or actual conflicts of interest, or the FDA concludes that the financial relationship may have affected interpretation of the
study, the integrity of the data generated at the applicable clinical trial site may be questioned and the utility of the clinical trial
itself may be jeopardized. The delay, suspension or discontinuation of any of our clinical trials, or a delay in the analysis of clinical
data for our product candidates, for any of the foregoing reasons, could adversely affect our efforts to obtain regulatory approval for
and to commercialize our product candidates, increase our operating expenses and have a material adverse effect on our financial results.
Clinical trials may also be
delayed or terminated as a result of ambiguous or negative interim results. In addition, a clinical trial may be suspended or terminated
by us, the FDA, the IRBs at the sites where the IRBs are overseeing a trial, or a data safety monitoring board, or DSMB (Data Safety Monitoring
Board)/DMC (Data Monitoring Committee), overseeing the clinical trial at issue, or other regulatory authorities due to a number of factors,
including:
| |
● |
failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
| |
● |
inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities resulting in the imposition of a clinical hold; |
| |
● |
varying interpretation of data by the FDA or similar foreign regulatory authorities; |
| |
● |
failure to achieve primary or secondary endpoints or other failure to demonstrate efficacy; |
| |
● |
unforeseen safety issues; or |
| |
● |
lack of adequate funding to continue the clinical trial. |
Modifications to our product candidates
may require federal approvals.
The BLA application is the
vehicle through which the company may formally propose that the FDA approve a new pharmaceutical for sale and marketing in the United
States. Once a particular product candidate receives FDA approval, expanded uses or uses in new indications of our products may require
additional human clinical trials and new regulatory approvals, including additional IND and BLA submissions and premarket approvals before
we can begin clinical development, and/or prior to marketing and sales. If the FDA requires new approvals for a particular use or indication,
we may be required to conduct additional clinical studies, which would require additional expenditures and harm our operating results.
If the products are already being used for these new indications, we may also be subject to significant enforcement actions.
Conducting clinical trials
and obtaining approvals is a time-consuming process, and delays in obtaining required future approvals could adversely affect our ability
to introduce new or enhanced products in a timely manner, which in turn would have an adverse effect on our business prospects, financial
condition and results of operation.
Clinical trials necessary to support approval
of our product candidates are time-consuming and expensive.
Initiating and completing
clinical trials necessary to support FDA approval of a BLA for Iomab-B, Actimab-A, and other product candidates, is a time-consuming and
expensive process, and the outcome is inherently uncertain. Moreover, the results of early clinical trials are not necessarily predictive
of future results, and any product candidate we advance into clinical trials may not have favorable results in later clinical trials.
For instance, we worked with
the FDA to develop the SIERRA clinical trial to test the safety and efficacy of Iomab-B in patients with r/r AML who are aged 55 and above
prior to a BMT. Even though the SIERRA trial met the primary endpoint of dCR with statistical significance (p-value<0.0001), the FDA
has determined that the analyses from the SIERRA trial do not support a BLA filing for Iomab-B. The FDA now requires an additional head-to-head
Phase 3 clinical study. We have further discussed the specifics of this additional clinical trial with the FDA. Based on these discussions,
Actinium believes it has aligned with the FDA on the patient population for this additional clinical trial, which can include all adult
patients aged 18 and above with active AML with blasts counts greater than 5% and less than 20%. This is a broader patient population
than the patients enrolled on the SIERRA trial, which only enrolled patients aged 55 and above. Further, the FDA now requires that an
additional dose optimization trial demonstrating safety and efficacy be completed to calculate the dose of Iomab-B based on absorbed dose
by the bone marrow, rather than the maximum tolerable dose of 24 Gy of radiation to the liver as was done in the SIERRA trial based on
several interactions we had with the FDA before starting the SIERRA trial. We are seeking a strategic partner for Iomab-B in the U.S.
to advance these additional trials. Even if we are able to secure a partner, there are no assurances that the additional trials will be
successful or that we can satisfy all of the FDA’s requests. There could be additional regulatory hurdles that may result in either
non-acceptance or non-approval of a future BLA filing which, in addition to clinical data, encompasses preclinical, CMC, labeling and
other information.
Preliminary, Interim, and “top-line”
data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject
to audit and verification procedures that could result in material changes in the final data.
From time to time, we may
publicly disclose preliminary, interim, and top-line data from our clinical trials, which is based on a preliminary analysis of then-available
data, and the results and related findings and conclusions are subject to change as more patient data become available or following a
more comprehensive review of the data related to the particular study or trial. We may also make assumptions, estimations, calculations
and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all
data. Our clinical trials may be open label studies and certain of our clinical development and or operations staff may review interim
or preliminary safety or efficacy data during routine data collection, cleaning and analysis from time to time. Interim or preliminary
results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such
results once additional data have been received and fully evaluated. Preliminary, interim or top-line data also remain subject to audit
and verification procedures that may result in the final data being materially different from the top-line, interim or preliminary data
we previously published. As a result, top-line, interim and preliminary data should be viewed with caution until the final data are available.
From time to time, we may
also disclose interim data from our preclinical studies and clinical trials. Interim data from clinical trials that we may complete are
subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data
become available. Adverse differences between interim data and final data could significantly harm our business prospects. Further, disclosure
of interim data by us or by our competitors could result in volatility in the price of our common stock.
Further, others, including
regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions, or analyses or may interpret
or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization
of the particular product candidate or product and our company in general. In addition, the information we choose to publicly disclose
regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with
what we determine is material or otherwise appropriate information to include in our disclosure.
If the interim, top-line or
preliminary data that we report differ from final results, or if others, including regulatory authorities, disagree with the conclusions
reached, our ability to obtain approval for, and commercialize, our product candidates may be harmed, which could harm our business, operating
results, prospects or financial condition.
Our clinical trials may fail to demonstrate
adequately the efficacy and safety of our product candidates, which would prevent or delay regulatory approval and commercialization.
Even if our clinical trials
are completed as planned, we cannot be certain that their results will support our product candidate claims or that the FDA or foreign
authorities will agree with our conclusions regarding them. Success in pre-clinical studies and early clinical trials does not ensure
that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials
and pre-clinical studies. The clinical trial process may fail to demonstrate that our product candidates are safe and effective for the
proposed indicated uses. If FDA concludes that the clinical trials for Iomab-B, Actimab-A, Iomab-ACT, or any other product candidate for
which we might seek approval, have failed to demonstrate safety and effectiveness, we would not receive FDA approval to market that product
candidate in the United States for the indications sought. In addition, such an outcome could cause us to abandon the product candidate
and might delay the development of others. Any delay or termination of our clinical trials will delay or preclude the filing of any submissions
with the FDA and, ultimately, our ability to commercialize our product candidates and generate revenues. It is also possible that patients
enrolled in clinical trials will experience adverse side effects that are not currently part of a product candidate’s profile.
The intellectual property related to antibodies
we have licensed has expired or likely expired.
The key patents related to
the humanized antibody, lintuzumab, which we use in our Actimab-A product candidate have expired. It is generally possible that others
may be eventually able to use an antibody with the same sequence, and we will then need to rely on additional patent protection covering
alpha particle drug products comprising Ac-225. Our final drug construct, Actimab-A, consists of the lintuzumab antibody labeled with
the isotope Ac-225. We currently own issued and pending patents relating to methods of manufacturing Actimab-A, methods of treatment using
Actimab-A and production of the Ac-225 isotope. In addition, we possess trade secrets and know how related to the manufacturing and use
of isotopes. Any competing product based on the lintuzumab antibody is likely to require several years of development before achieving
our product candidate’s current status and may be subject to significant regulatory hurdles but such development by others is nevertheless
a possibility that could negatively impact our business in the future. We own 4 issued U.S. patents, 2 issued Canadian patents, 1 issued
European patent (validated as a national patent in several countries) and 1 issued Japanese patent that relate to the composition of our
Iomab-B product candidate. Patent applications relating to Iomab-B are also pending in the U.S. and internationally. We have and may continue
to file patents related to Iomab-B that can provide barriers to entry but there is no certainty that these patents will be granted or
such granting thereof will adequately prevent others from seeking to replicate and use the apamistamab antibody or the construct. Our
patent portfolio includes pending applications related to radioimmunoconjugate composition, formulation administration, and methods of
use in treating solid or liquid cancers. This subject matter includes composition, administration, and methods of treatment for our product
candidates Actimab-A and Iomab-B. Any competing product based on the antibody used in Iomab-B is likely to require several years of development
before achieving our product candidate’s current status and may be subject to significant regulatory hurdles. Further, if approved,
Iomab-B would be entitled to 12 years of market exclusivity in the U.S. and 10 years in Europe, during which time no generic biologic
or biosimilar referencing Iomab-B can be granted marketing approval.
Our CD33 program clinical trials are testing
the same drug construct.
Our CD33 program is expected
to be comprised of several clinical trials conducted under the CRADA with NCI, Actinium sponsored trials, or investigator-initiated trials
in AML and other myeloid indications that will study the same drug construct consisting of lintuzumab-Ac-225. Negative results from any
of these trials could negatively impact our ability to enroll or complete our other trials studying lintzumab-Ac-225, including future
studies conducted under our CRADA with the NCI. Additionally, negative outcomes including safety concerns, may result in the FDA requiring
amendment to certain clinical trials, placing a clinical hold on certain or all clinical trials or discontinuing other trials utilizing
lintuzumab-Ac-225.
We may be unable to obtain a sufficient
supply of isotopes to support clinical development or at commercial scale.
Iodine-131 is a key component
of our Iomab-B drug candidate. We source medical grade I-131 from multiple suppliers, including two leading global manufacturers. Currently,
we believe there is sufficient supply of I-131 to support additional trials we may undertake utilizing I-131 and for future commercialization
of potential I-131 based products. We continually evaluate I-131 manufacturers and suppliers. While we consider I-131 to be commoditized
and obtainable through several suppliers, there can be no guarantee that we will be able to secure I-131 or obtain I-131 on terms that
are acceptable to us.
Actinium-225 is a key component of our Actimab-A product candidate,
technology platform, preclinical R&D programs and other drug candidates that we might consider for development with the Ac-225 payload.
We have secured multiple suppliers that are expected to provide cGMP Ac-225 for our planned clinical trials. There are adequate quantities
of Ac-225 available today to meet our current needs via our present supplier, the Department of Energy (“DOE”), who has been
our primary supplier of Ac-225 historically. The Ac-225 currently supplied for our clinical trials from the DOE is derived from the natural
decay of thorium-229 from so-called ‘thorium-cows’ and is able to produce sufficient quantities that are several multiples
of the amount of Ac-225 we require to supply our clinical programs through to early commercialization phase. The DOE is also producing
Ac-225 from a recently developed alternative route for Ac-225 production via a linear accelerator that is currently being evaluated by
us. Initial preclinical and modelling results have indicated that the linear accelerator sourced Ac-225 does not impact labelling efficiency
and expected distribution. In accordance with representations made by the DOE, the capacity of Ac-225 from this route is expected to be
sufficient to supply all of Actinium’s pipeline and commercial Ac-225 needs and support new program expansion by not just Actinium
but also other companies that are developing Ac-225 based products. Additional routes of Ac-225 production are being pursued by the DOE
including the generation of new thorium cows and production via a cyclotron. The cyclotron production method for Ac-225 production leverages
Actinium’s proprietary technology and know-how and presents an additional path towards production of high-quality Ac-225 at a scale
that would be able to satisfy commercial needs. In addition, we are aware of at least ten other government and non-government entities
globally including the U.S., Canada, Russia, Belgium, France and Japan that have, or expect to have ability to supply Ac-225 or equipment
for its production within the timeframes relevant to the potential first commercial approval of our Ac-225-based drug candidate.
Our contract for supply of this isotope from the DOE must be renewed
yearly, and we renewed our contract to extend through the end of 2024. While we expect this contract will continue to be renewed at the
end of its term as it has since 2009, there can be no assurance that the DOE will renew the contract or that change its policies that
allow for the sale of isotope to us. There can be no assurance that the DOE or our other suppliers will be able to supply all of the quantities
of Ac-225 we request in the future. Failure to acquire sufficient quantities of medical grade Ac-225 would make it impossible to effectively
complete clinical trials and to commercialize any Ac-225 based drug candidates that we may develop and would materially harm our business.
Our ability to conduct clinical
trials to advance our drug candidates is dependent on our ability to obtain the radioisotopes I-131, Ac-225 and other isotopes we may
choose to utilize in the future. Currently, we are dependent on third party manufacturers and suppliers for our isotopes. These suppliers
may not perform their contracted services or may breach or terminate their agreements with us. Our suppliers are subject to regulations
and standards that are overseen by regulatory and government agencies and we have no control over our suppliers’ compliance to these
standards. Failure to comply with regulations and standards may result in their inability to supply isotopes and could result in delays
in our clinical trials, which could have a negative impact on our business. We have developed intellectual property, know-how and trade
secrets related to the manufacturing process of Ac-225. While we have manufactured medical grade Ac-225 of a purity compared to the cyclotron
sourced material in the past, this activity was terminated due to operating cost reasons, and we currently do not have experience in manufacturing
medical grade Ac-225 and may not obtain the resources necessary to establish our own manufacturing capabilities in the future. Our inability
to build out and establish our own manufacturing facilities would require us to continue to rely on third party suppliers as we currently
do. However, based on our current third-party suppliers and potential future suppliers of Ac-225 we expect to have adequate isotope supply
to support our current ongoing clinical trials, current and planned preclinical R&D activities and commercialization should our drug
candidates receive regulatory approval.
If we encounter difficulties enrolling patients
in our clinical trials, our clinical development activities could be delayed or otherwise adversely affected.
The timely completion of clinical
trials in accordance with their protocols depends on our ability to enroll a sufficient number of patients who remain in the trial until
its conclusion. We may experience difficulties in patient enrollment in our clinical trials for a variety of reasons, including:
| |
● |
the size and nature of the patient population; |
| |
● |
the patient eligibility criteria defined in the protocol; |
| |
● |
the size of the study population required for analysis of the trial’s primary endpoints; |
| |
● |
the proximity of patients to trial sites; |
| |
● |
the design of the trial; |
| |
● |
our ability to recruit clinical trial investigators with the appropriate competencies and expertise; |
| |
● |
competing clinical trials for similar or alternate therapeutic treatments; |
| |
● |
clinician’s and patients’ perceptions as to the potential advantages and side effects of the product candidate being studied in relation to other available therapies; |
| |
● |
our ability to obtain and maintain patient consents; and |
| |
● |
the risk that patients enrolled in clinical trials will not complete a clinical trial. |
In addition, refractory patients, which several of our trials have
or are expected to enroll, participating in clinical trials are seriously and often terminally ill and therefore may not complete the
clinical trial due to reasons including comorbid conditions or occurrence of adverse medical events related or unrelated to the investigational
products, or death. Even if we are able to enroll a sufficient number of patients in our clinical trials, delays in patient enrollment
will result in increased costs or affect the timing of our planned trials, which could adversely affect our ability to advance the development
of our product candidates.
FDA may take actions that would prolong,
delay, suspend, or terminate clinical trials of our product candidates, which may delay or prevent us from commercializing our product
candidates on a timely basis.
There can be no assurance
that the data generated in our clinical trials will be acceptable to FDA or that if future modifications during the trial are necessary,
that any such modifications will be acceptable to FDA. Certain modifications to a clinical trial protocol made during the course of the
clinical trial have to be submitted to the FDA. This could result in the delay or halt of a clinical trial while the modification is evaluated.
In addition, depending on the quantity and nature of the changes made, FDA could take the position that some or all of the data generated
by the clinical trial is not usable because the same protocol was not used throughout the trial. This might require the enrollment of
additional subjects, which could result in the extension of the clinical trial and the FDA delaying approval of a product candidate. If
the FDA believes that its prior approval is required for a particular modification, it can delay or halt a clinical trial while it evaluates
additional information regarding the change.
Any delay or termination of
our current or future clinical trials as a result of the risks summarized above, including delays in obtaining or maintaining required
approvals from IRBs, delays in patient enrollment, the failure of patients to continue to participate in a clinical trial, and delays
or termination of clinical trials as a result of protocol modifications or adverse events during the trials, may cause an increase in
costs and delays in the filing of any submissions with the FDA, delay the approval and commercialization of our product candidates or
result in the failure of the clinical trial, which could adversely affect our business, operating results and prospects. Lengthy delays
in obtaining regulatory approval for Iomab-B or completion of our ongoing or planned clinical trials would adversely affect our business
and prospects and could cause us to cease operations.
We have obtained orphan drug designation
from FDA for two of our current product candidates and intend to pursue such designation for other candidates and indications in the future,
but we may be unable to obtain such designations or to maintain the benefits associated with any orphan drug designations we have received
or may receive in the future.
We have received orphan drug
designation for Iomab-B and Actimab-A for treatment of AML in both the United States and the EU. Under the Orphan Drug Act, the FDA may
grant orphan designation to a drug or biologic intended to treat a rare disease or condition, which is a disease or condition that affects
fewer than 200,000 individuals in the United States, or if it affects more than 200,000 individuals in the United States, there is no
reasonable expectation that the cost of developing and making available a drug or biologic for this type of disease or condition will
be recovered from sales in the United States for that drug or biologic. Similarly, the EMA grants orphan drug designation to promote the
development of products that are intended for the diagnosis, prevention, or treatment of a life-threatening or chronically debilitating
condition affecting not more than five in 10,000 persons in the EU.
Orphan drug designation neither
shortens the development time or regulatory review time of a drug or biologic nor gives the drug or biologic any advantage in the regulatory
review or approval process. In the United States, orphan drug designation entitles a party to financial incentives, such as opportunities
for grant funding towards clinical trial costs, tax advantages, and application fee waivers. In addition, if a product candidate receives
the first FDA approval for the indication for which it has orphan designation, such product is entitled, upon approval, to seven years
of orphan-drug exclusivity, during which the FDA may not approve any other application to market the same drug for the same indication,
unless a subsequently approved product is clinically superior to orphan drug or where the manufacturer is unable to assure sufficient
product quantity in the applicable patient population. In the EU, orphan drug designation entitles a party to financial incentives such
as reduction of fees or fee waivers and ten years of market exclusivity following drug or biological product approval. This period may
be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently
profitable not to justify maintenance of market exclusivity.
Even if we obtain (or have
obtained) orphan drug designation for certain product candidates, we may not be the first to obtain marketing approval for such candidates
for the applicable indications due to the uncertainties inherent in the development of novel biologic products, and, an orphan drug candidate
may not receive orphan-drug exclusivity upon approval if such candidate is approved for a use that is broader than the indication for
which it received orphan designation. In addition, exclusive marketing rights in the United States may be lost if the FDA later determines
that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product
to meet the needs of patients with the rare disease or condition.
Finally, even if we successfully
obtain orphan-drug exclusivity for an orphan drug candidate upon approval, such exclusivity may not effectively protect the product from
competition because (i) different drugs with different active moieties can be approved for the same condition; and (ii) the FDA or EMA
can also subsequently approve a subsequent product with the same active moiety and for the same indication as the orphan drug if the later-approved
drug if deemed clinically superior to the orphan drug.
Even if we receive regulatory approval of
our product candidates, we will be subject to ongoing regulatory obligations and continued regulatory review.
Any regulatory approvals that
we receive for our product candidates will require surveillance to monitor the safety and efficacy of the product candidate. The FDA may
also require a REMS in order to approve our product candidates, which could entail requirements for a medication guide, physician communication
plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization
tools. In addition, if the FDA or a comparable foreign regulatory authority approves our product candidates, the manufacturing processes,
labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, import, export and recordkeeping for our
product candidates will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety
and other post-marketing information and reports, registration, as well as continued compliance with cGMPs and GCPs for any clinical trials
that we conduct post-approval. In addition, the FDA could require us to conduct another study to obtain additional safety or biomarker
information. Later discovery of previously unknown problems with our product candidates, including adverse events of unanticipated severity
or frequency, or with our third-party suppliers or manufacturing processes, or failure to comply with regulatory requirements, may result
in, among other things:
| |
● |
restrictions on the marketing or manufacturing of our product candidates, withdrawal of the product from the market, or voluntary or mandatory product recalls; |
| |
● |
fines, warning letters or holds on clinical trials; |
| |
● |
refusal by the FDA to approve pending applications or supplements to approved applications filed by us or suspension or revocation of license approvals; |
| |
● |
product seizure or detention, or refusal to permit the import or export of our product candidates; and |
| |
● |
injunctions or the imposition of civil or criminal penalties. |
The FDA’s and other
regulatory authorities’ policies may change, and additional government regulations may be enacted that could prevent, limit or delay
regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise
from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes
in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we
may lose any marketing approval that we may have obtained, and we may not achieve or sustain profitability.
Coverage and reimbursement may be limited
or unavailable in certain market segments for our product candidates which could limit our sales of our product candidates, if approved.
The commercial success of
our product candidates in both domestic and international markets will be substantially dependent on whether third-party coverage and
reimbursement is available for patients that use our products. However, the availability of insurance coverage and reimbursement for newly
approved cancer therapies is uncertain, and therefore, third-party coverage may be particularly difficult to obtain even if our products
are approved by the FDA as safe and efficacious. Patients using existing approved therapies are generally reimbursed all or part of the
product cost by Medicare or other third-party payors. Medicare, Medicaid, health maintenance organizations and other third-party payors
are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement of new drugs, and, as
a result, they may not cover or provide adequate payment for these products. Submission of applications for reimbursement approval generally
does not occur prior to the filing of a BLA for that product and may not be granted until many months after BLA approval. In order to
obtain coverage and reimbursement for these products, we or our commercialization partners may have to agree to a net sales price lower
than the net sales price we might charge in other sales channels. The continuing efforts of government and third-party payors to contain
or reduce the costs of healthcare may limit our revenue. Initial dependence on the commercial success of our products may make our revenues
particularly susceptible to any cost containment or reduction efforts.
Healthcare legislative reform measures intended
to increase pressure to reduce prices of pharmaceutical products paid for by Medicare or, otherwise, affect the federal regulation of
the U.S. healthcare system could have a material adverse effect our business, future revenue, if any, and results of operations.
In the United States, there
have been a number of legislative and regulatory initiatives focused on containing the cost of healthcare. The Affordable Care Act, for
example, substantially changed the way healthcare is financed by both governmental and private insurers. The Affordable Care Act contains
a number of provisions that could impact our business and operations, primarily, once we obtain FDA approval to commercialize one of our
product candidates in the United States, if ever, and may also affect our operations in ways we cannot currently predict. Affordable Care
Act provisions that may affect our business include, among others, those governing enrollment in federal healthcare programs, reimbursement
changes, rules regarding prescription drug benefits under health insurance exchanges, expansion of the 340B program, expansion of state
Medicaid programs, fees and increased discount and rebate obligations, transparency and reporting requirements, and fraud and abuse enforcement.
Such changes may impact existing government healthcare programs, industry competition, formulary composition, and may result in the development
of new programs, including Medicare payment for performance initiatives, health technology assessments, and improvements to the physician
quality reporting system and feedback program.
There have been significant
judicial, administrative, executive, and legislative initiatives to modify, limit, replace, or repeal the Affordable Care Act since its
enactment. For example, former President Trump issued several Executive Orders and other directives designed to delay the implementation
of certain provisions of the Affordable Care Act or otherwise circumvent some of the requirements for health insurance mandated by the
Affordable Care Act. Concurrently, Congress considered legislation that would repeal or replace all or part of the Affordable Care Act.
While Congress has not passed comprehensive repeal legislation, several bills affecting the implementation the Affordable Care Act have
been passed. For example, the Tax Cuts and Jobs Act of 2017 eliminated the Affordable Care Act provision requiring individuals to purchase
and maintain health coverage, or the “individual mandate,” by reducing the associated penalty to zero, beginning in 2019.
In December 2018, a district court in Texas held that the individual mandate is unconstitutional and that the rest of the Affordable Care
Act is, therefore, invalid. On appeal, the Fifth Circuit Court of Appeals affirmed the holding on the individual mandate but remanded
the case back to the lower court to reassess whether and how such holding affects the validity of the rest of the Affordable Care Act.
The Fifth Circuit’s decision on the individual mandate was appealed to the U.S. Supreme Court. On June 17, 2021, the Supreme Court
held that the plaintiffs (comprised of the state of Texas, as well as numerous other states and certain individuals) did not have standing
to challenge the constitutionality of the Affordable Care Act’s individual mandate and, accordingly, vacated the Fifth Circuit’s
decision and instructed the district court to dismiss the case. As a result, the Affordable Care Act will remain in-effect in its current
form for the foreseeable future; however, we cannot predict what additional challenges may arise in the future, the outcome thereof, or
the impact any such actions may have on our business.
In addition to the Affordable
Care Act, there have been numerous other Congressional initiatives and proposed and enacted federal and state legislation designed to,
among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs,
and reform government program reimbursement methodologies for drug products. Pharmaceutical product prices have been the focus of increased
scrutiny by the government, including certain state attorneys general, members of Congress and the United States Department of Justice.
State or federal healthcare reform measures or other social or political pressure to lower the cost of pharmaceutical products could have
a material adverse impact on our business, results of operations and financial condition.
The Biden administration also
introduced various measures in 2021 focusing on healthcare and drug pricing, in particular. For example, on January 28, 2021, President
Biden issued an executive order that initiated a special enrollment period for purposes of obtaining health insurance coverage through
the Affordable Care Act marketplace, which began on February 15, 2021, and remained open through August 15, 2021. The executive order
also instructed certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare,
including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements and policies that
create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the Affordable Care Act. On the legislative
front, the American Rescue Plan Act of 2021 was signed into law on March 11, 2021, which, in relevant part, eliminates the statutory Medicaid
drug rebate cap, currently set at 100% of a drug’s average manufacturer price, for single source drugs and innovator multiple source
drugs, beginning January 1, 2024. And, in July 2021, the Biden administration released an executive order entitled, “Promoting Competition
in the American Economy,” with multiple provisions aimed at prescription drugs. In response, on September 9, 2021, HHS released
a “Comprehensive Plan for Addressing High Drug Prices” that outlines principles for drug pricing reform and sets out a variety
of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can take to advance these
principles.
More recently, on August 16,
2022, President Biden signed into law the Inflation Reduction Act of 2022 (the “IRA”), which, among other
provisions, included several measures intended to lower the cost of prescription drugs and related healthcare reforms. Specifically, the
IRA authorizes and directs the Department of Health and Human Services (the “DHHS”) to set drug price caps for certain high-cost
Medicare Part B and Part D qualified drugs, with the initial list of drugs announced on August 29, 2023, and the first year of maximum
price applicability to begin in 2026. The IRA further authorizes the DHHS to penalize pharmaceutical manufacturers that increase the price
of certain Medicare Part B and Part D drugs faster than the rate of inflation. Finally, the IRA creates significant changes to the Medicare
Part D benefit design by capping Part D beneficiaries’ annual out-of-pocket spending at $2,000 beginning in 2025. We cannot be sure
whether additional or related legislation or rulemaking will be issued or enacted, or what impact, if any, such changes will have on the
profitability of any of our drug candidates, if approved for commercial use, in the future.
Our relationships with customers, health
care professionals and third-party payors may be subject to applicable healthcare laws, which could expose us to penalties, including
administrative, civil or criminal penalties, damages, fines, imprisonment, exclusion from participation in federal healthcare programs
such as Medicare and Medicaid, reputational harm, the curtailment or restructuring of our operations and diminished future profits and
earnings.
Healthcare professionals and
third-party payors will play a primary role in the recommendation and prescription of any product candidates for which we obtain marketing
approval. Our current and future arrangements with customers, healthcare professionals and third-party payors may expose us to broadly
applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships
through which we conduct research, market, sell and distribute any products for which we obtain marketing approval. Federal and state
healthcare laws and regulations that may affect our operations, directly or indirectly, include the following, among others:
| |
● |
the federal Anti-Kickback Statute, which prohibits persons and entities from, among other things, knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, lease, order or recommendation of, any good, facility, item or service, for which payment may be made under federal and state healthcare programs such as Medicare and Medicaid; |
| |
● |
the federal false claims laws, including civil whistleblower or qui tam actions under the federal False Claims Act, which impose criminal and civil penalties against individuals or entities for, among other things, knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; |
| |
● |
the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, which imposes criminal and civil liability for, among other things, executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters and also imposes obligations, including mandatory contractual terms, on covered entities, including certain healthcare providers, health plans, and healthcare clearinghouses, and their respective business associates that create, receive, maintain or transmit individually identifiable health information for or on behalf of the covered entity as well as their covered subcontractors, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| |
● |
the federal Civil Monetary Penalties Law, which prohibits, among other things, the offering or transfer of remuneration to a Medicare or state healthcare program beneficiary if the person knows or should know it is likely to influence the beneficiary’s selection of a particular provider, practitioner, or supplier of services reimbursable by Medicare or a state healthcare program, unless an exception applies; |
| |
● |
the federal Physician Payments Sunshine Act, created under the Affordable Care Act, and its implementing regulations, which requires certain manufacturers of drugs, devices, biologicals and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually information related to certain payments or other transfers of value provided to physicians and any ownership and investment interests held by physicians or their immediate family members. Beginning in 2022, applicable manufacturers also will be required to report such information regarding payments and other transfers of value to physician assistants, nurse practitioners, clinical nurse specialists, anesthesiologist assistants, certified registered nurse anesthetists and certified nurse midwives during the previous year; and |
| |
● |
analogous state laws and regulations, including (among others) state anti-kickback and false claims laws, which may apply to our business practices, including, but not limited to, research, distribution, sales and marketing arrangements and claims involving healthcare items or services reimbursed by any third-party payor, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the United States federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws and regulations that require drug manufacturers to file reports relating to pricing and marketing information and that require tracking gifts and other remuneration and items of value provided to healthcare professionals and entities; state and local laws that require the registration of pharmaceutical sales representatives; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by federal law, thus complicating compliance efforts. |
Efforts to comply with applicable
healthcare laws and regulations will involve substantial costs. Interpretations of standards of compliance under these laws and regulations
are rapidly changing and subject to varying interpretations and it is possible that governmental authorities will conclude that our business
practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare
laws and regulations. If our operations are found to be in violation of any of these laws or any other laws that may apply to us, we may
be subject to significant civil, criminal and administrative penalties, damages, fines, exclusion from government funded healthcare programs,
such as Medicare and Medicaid, reputational harm, imprisonment, additional reporting obligations and oversight (if we become subject to
a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws), and the curtailment or restructuring
of our operations, any of which could diminish our future profits or earnings. If any of the physicians or other providers or entities
with whom we expect to do business are found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative
sanctions, including exclusions from government funded healthcare programs.
Third-party payors may not adequately reimburse
customers for any product candidates that we may commercialize or promote, and may impose coverage restrictions or limitations such as
prior authorizations and step edits that affect their use.
Our ability to commercialize
any product candidates successfully also will depend in part on the extent to which coverage and adequate reimbursement for these products
and related treatments will be available from government health programs, private health insurers, integrated delivery networks and other
third-party payors. Third-party payors decide which medications they will pay for and establish reimbursement levels. A significant trend
in the United States healthcare industry and elsewhere is cost containment. Government authorities and third-party payors have attempted
to control costs by limiting coverage and the amount of payment for particular medications. Increasingly, third-party payors are requiring
that drug companies provide predetermined discounts from list prices and are challenging the prices charged for medical products. Coverage
and reimbursement may not be available for any product that we commercialize and, if reimbursement is available, the level of reimbursement
may not be sufficient for commercial success. Coverage and reimbursement may impact the demand for, or the price of, any product candidate
for which we obtain marketing approval. If coverage and reimbursement is not available or is available only to limited levels, we may
not be able to successfully commercialize any product candidate for which we obtain marketing approval.
Obtaining reimbursement approval
for any product candidate for which we obtain marketing approval from any government or other third-party payor is a time-consuming and
costly process. There may be significant delays in obtaining coverage and adequate reimbursement for newly approved products. Moreover,
eligibility for coverage and reimbursement does not imply that any product will be paid for in all cases or at a rate that covers our
costs, including research, development, manufacture, sale and distribution. Even when a payor determines that a product that we may commercialize
or promote is eligible for reimbursement under its criteria, the payor may impose coverage limitations that preclude payment for some
uses that are approved by the FDA, or may impose restrictions, such as prior authorization requirements, or may simply deny coverage altogether.
Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent.
Coverage and reimbursement rates may vary according to the use of the drug and the medical circumstances under which it is used may be
based on reimbursement levels already set for lower cost products or procedures or may be incorporated into existing payments for other
services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private
payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices
than in the United States. Furthermore, the Centers for Medicare and Medicaid Services frequently change product descriptors, coverage
policies, product and service codes, payment methodologies and reimbursement values. Commercial third-party payors often rely upon Medicare
coverage policies and payment limitations in setting their own reimbursement policies. Our inability to promptly obtain and maintain coverage
and profitable payment rates from both government-funded programs and private payors for any approved products that we develop could have
a material adverse effect on our operating results, our ability to raise capital needed to commercialize our approved products and our
overall financial condition.
Risks Related to Third Parties
We may rely on third parties to conduct
our clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines or comply
with regulatory requirements, we may not be able to obtain regulatory approval for or commercialize our product candidates.
We do not have the ability
to independently conduct our clinical trials for our product candidates and we must rely on third parties, such as contract research organizations,
medical institutions, clinical investigators and contract laboratories to conduct such trials. Our reliance on these third parties for
clinical development activities results in reduced control over these activities. Moreover, the FDA requires us to comply with regulations
and standards, commonly referred to as GCPs (good clinical practices), for conducting, recording and reporting the results of clinical
trials to assure that data and reported results are credible and accurate and that the trial participants are adequately protected. Our
reliance on third parties does not relieve us of these responsibilities and requirements. If we or any of our third-party contractors
fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable
foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot
assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials
complies with GCP regulations. In addition, our clinical trials must be conducted with product produced under current good manufacturing
practice, or cGMP, regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay
the regulatory approval process.
If our consultants, contract
research organizations and other similar entities with which we are working do not successfully carry out their contractual duties, meet
expected deadlines, or comply with applicable regulations, we may be required to replace them. Although we believe that there are a number
of other third-party contractors we could engage to continue these activities, we may not be able to enter into arrangements with alternative
third-party contractors or to do so on commercially reasonable terms, which may result in a delay of our planned clinical trials and delayed
development of our product candidates.
In addition, our third-party
contractors are not our employees, and except for remedies available to us under our agreements with such third-party contractors, we
cannot control whether or not they devote sufficient time and resources to our programs. If these third parties do not successfully carry
out their contractual duties or regulatory obligations or meet expected deadlines, or if the quality or accuracy of the data they obtain
is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our pre-clinical
development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory
approval for, or successfully commercialize, our product candidates on a timely basis, if at all, and our business, operating results
and prospects would be adversely affected.
The protection against generic competition
for our biologic drug candidates and reimbursement by CMS may be subject to future change
We are not aware of any existing
or pending regulations or legislation that pertains to generic radiopharmaceutical products such as our ARC targeted radiotherapy product
candidates. Our ARC product candidates are regulated by the FDA as biologic products, and we intend to seek approval for these products
pursuant to the BLA pathway. The Biologics Price Competition and Innovation Act of 2009, or BPCIA, created an abbreviated pathway for
the approval of biosimilar and interchangeable biologic products. The abbreviated regulatory pathway establishes legal authority for the
FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based
on its similarity to an existing brand product. Under the BPCIA, an application for a biosimilar product cannot be approved by the FDA
until 12 years after the original branded product was approved under a BLA and in Europe a biosimilar product cannot be approved until
10 years after the original branded product was approved. The law is complex and as a result, its ultimate impact, implementation, and
meaning are subject to uncertainty. Even if a biosimilar gets approved for one of the antibodies that we use, the final constructs of
our drug candidates consist of an antibody, radioisotope and in some cases a linker and we are not aware of any regulations that would
require us to provide the final constructs or components to third parties or potential competitors. Therefore, based on the current regulations,
we do not believe that the final drug product of our candidates can be subject to competition from a biosimilar as outlined in BPCIA for
at least 12 years in the U.S. and 10 years in the EU. We are aware that generic versions of certain radiopharmaceuticals utilizing peptides
have been submitted to the FDA via the Abbreviated New Drug Application (“ANDA”) pathway, however, those products are not
covered under the BPCIA and therefore that generic pathway is not applicable to Iomab-B or Actimab-A. We expect this would also apply
to other biologic drug candidates we may seek to develop in the future based on the current provisions of the BPCIA. Additionally, the
Inflation Reduction Act (“IRA”) that was enacted in August 2022, states that reimbursement by the Centers for Medicare&
Medicaid Services (“CMS”) for high-expenditure single-source biologic drugs, which we expect Iomab-B and Actimab-A to be,
can only be negotiated after at least 11 years following approval compared to 7 years for non-biologic drugs with negotiated prices taking
effect two years after selection. Therefore, we currently believe that our ARCs are less likely than small molecules to face pricing pressure
and negotiation from IRA. Further, a drug or biological product that has an orphan drug designation, which Iomab-B and Actimab-A both
have, for only one rare disease or condition will be excluded from the IRA’s price negotiations requirements until such time
the biological products has designations for more than one rare disease or condition, or if is approved for an indication that is not
within that single designated rare disease or condition, unless such additional designation or such disqualifying approvals are withdrawn
by the time CMS evaluates the drug for selection for negotiation. In August 2023, 10 initial drugs were identified with negotiated prices
expected to take effect starting in 2026. In 2027 and 2028, it is expected that CMS will establish negotiated prices for 15 additional
drugs in each respective year. We do not believe there is a high likelihood that Iomab-B or Actimab-A would be identified by CMS for negotiated
pricing under IRA but there is potential that IRA and other additional state and federal healthcare reform measures will be adopted in
the future and the implementation of cost-containment measures or other healthcare reforms may prevent us from being able to generate
revenue, attain profitability or successfully commercialize our product candidates.
Our product candidates may never achieve
market acceptance.
Iomab-B, Actimab-A, Iomab-ACT and future product candidates that we
may develop may never gain market acceptance among physicians, patients and the medical community. The degree of market acceptance of
any of our products will depend on a number of factors, including the actual and perceived effectiveness and reliability of the product;
the results of any long-term clinical trials relating to use of the product; the availability, relative cost and perceived advantages
and disadvantages of alternative technologies; the degree to which treatments using the product are approved for reimbursement by public
and private insurers; the strength of our marketing and distribution infrastructure; and the level of education and awareness among physicians
and hospitals concerning the product.
We believe that oncologists
and other physicians will not widely adopt a product candidate unless they determine, based on experience, clinical data, and published
peer-reviewed journal articles, that the use of that product candidate provides an effective alternative to other means of treating specific
cancers. Patient studies or clinical experience may indicate that treatment with our product candidates does not provide patients with
sufficient benefits in extension of life or quality of life. We believe that recommendations and support for the use of each product candidate
from influential physicians will be essential for widespread market acceptance. Our product candidates are still in the development stage,
and it is premature to attempt to gain support from physicians at this time. We can provide no assurance that such support will ever be
obtained. If our product candidates do not receive such support from these physicians and from long-term data, physicians may not use
or continue to use, and hospitals may not purchase or continue to purchase, them.
Failure of Iomab-B, Actimab-A, Iomab-ACT or any of our other product
candidates to significantly penetrate current or new markets would negatively impact our business financial condition and results of operations.
We may be subject to claims that our third-party
service providers, consultants or current or former employees have wrongfully used or disclosed confidential information of third parties.
We have received confidential
and proprietary information from third parties. In addition, we employ individuals who were previously employed at other biotechnology
or pharmaceutical companies. We may be subject to claims that we or our employees, consultants or independent contractors have inadvertently
or otherwise used or disclosed confidential information of these third parties or our employees’ former employers. Litigation may
be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial
cost and be a distraction to our management and employees.
We currently depend on single third-party
manufacturers to produce our pre-clinical and clinical trial drug supplies. Any disruption in the operations of our current third-party
manufacturers, or other third-party manufacturers we may engage in the future, could adversely affect our business and results of operations.
We do not currently operate
manufacturing facilities for pre-clinical or clinical production of any of our product candidates. We rely on third-party manufacturers
to supply, store, and distribute pre-clinical and clinical supply of the components of our drug product candidates including monoclonal
antibodies, linkers and radioisotopes, as well as the final construct which comprises our drug product candidates. We expect to continue
to depend on third-party manufacturers for the foreseeable future. Any performance failure on the part of our existing or future manufacturers
could delay clinical development, cause us to suspend or terminate development or delay or prohibit regulatory approval of our product
candidates or commercialization of any approved products. Further avenues of disruption to our clinical or eventual commercial supply
may also occur due to the sale, acquisition, business reprioritization, bankruptcy or other unforeseen circumstances that might occur
at any of our suppliers or contract manufacturing partners including an inability to come to terms on renewal of existing contracts or
new contracts.
We currently rely on single
manufacturers to manufacture our pre-clinical and clinical trial drug supplies. With a view to maintaining business continuity we are
evaluating alternatives and second and even third sources of supply or manufacturing for our core suppliers and manufacturing partners,
however there can be no assurances that we will be able to identify such suppliers or partners and assuming we did, that we would be able
to enter into contracts that are on favorable terms or on terms that will enable sufficient supply to ensure business continuity and support
our growth plans.
Our product candidates require
precise, high-quality manufacturing. Failure by our current contract manufacturer or other third-party manufacturers we may engage in
the future to achieve and maintain high manufacturing standards could result in patient injury or death, product recalls or withdrawals,
delays or failures in testing or delivery, cost overruns, or other problems that could seriously hurt our business. Contract manufacturers
may encounter difficulties involving production yields, quality control, and quality assurance. These manufacturers are subject to ongoing
periodic and unannounced inspections by the FDA and corresponding state and foreign agencies to ensure strict compliance with cGMPs and
other applicable government regulations and corresponding foreign standards; we do not have control over third-party manufacturers’
compliance with these regulations and standards.
We may elect to build or purchase
a manufacturing facility or facilities in the future to operate for the purposes of manufacturing our own products. We have never built,
owned or operated a manufacturing facility. There can be no assurances that we will be able to successfully accomplish this and in doing
so we may experience delays, cost overruns, or other problems that could seriously hurt our business. Even if we successfully build or
purchase a manufacturing facility, we may not realize the expected benefits of these efforts.
We depend on vendors with specialized operations, equipment and know-how
to manufacture the respective components of our drug candidates. We have entered into manufacturing and supply agreements with these third-parties,
and in some instances, we have agreed that such vendor be the exclusive manufacturer and supplier. If any of the third-parties we depend
on encounter difficulties in their operations, fail to comply with required regulations or breach their contractual obligations it may
be difficult, or we may be unable to identify suitable alternative third-party manufacturers. While we identify and evaluate third-party
manufacturers from time to time, even if we do identify suitable alternative third-parties, we may fail to reach agreement on contractual
terms, it may be prohibitively expensive and there can be no assurance that we can successfully complete technology transfer and development
work necessary, or complete the necessary work in a timely manner. Any of which could prevent us from commencing manufacturing with third-parties
which could cause delays or suspension of our clinical trials and pre-clinical work that may have a negative impact on our business.
Furthermore, these third-party
contractors, whether foreign or domestic, may experience regulatory compliance difficulty, mechanical shutdowns, employee strikes, or
any other unforeseeable acts that may delay or limit production. Our inability to adequately establish, supervise and conduct (either
ourselves or through third parties) all aspects of the formulation and manufacturing processes, and the inability of third-party manufacturers
to consistently supply quality product when required would have a material adverse effect on our ability to develop or commercialize our
products. We have faced delays and risks associated with reliance on key third party manufacturers in the past and may be faced with such
delays and risks in the future. Any future manufacturing interruptions or related supply issues could have an adverse effect on our company,
including delays in clinical trials.
If we are successful in obtaining marketing
approval from the FDA and/or other regulatory agencies for any of our product candidates, we anticipate continued reliance on third-party
manufacturers.
To date, our product candidates
have been manufactured in small quantities for preclinical and clinical testing by third-party manufacturers. If the FDA or other regulatory
agencies approve any of our product candidates for commercial sale, we expect that we would continue to rely, at least initially, on third-party
specialized manufacturers to produce commercial quantities of approved products. These manufacturers may not be able to successfully increase
the manufacturing capacity for any approved product in a timely or economic manner, or at all. Significant scale-up of manufacturing may
require additional validation studies, which the FDA must review and approve. Scale-up for commercial product may require financial commitment
or investment by us, which we may not have sufficient capital for or may elect not to undertake. If third party manufacturers are unable
to successfully increase the manufacturing capacity for a product candidate, or we are unable to establish our own manufacturing capabilities,
the commercial launch of any approved products may be delayed or there may be a shortage in supply, which in turn could have a material
adverse effect on our business.
In addition, the facilities
used by our contract manufacturers to manufacture our product candidates must be approved by the FDA pursuant to inspections that will
be conducted after we submit a BLA to the FDA. We do not control the manufacturing process of, and are completely dependent on, our contract
manufacturing partners for compliance with cGMPs. If our contract manufacturers cannot successfully manufacture material that conforms
to our specifications and the strict regulatory requirements of the FDA or other regulatory authorities, they will not be able to secure
and/or maintain regulatory approval for their manufacturing facilities. If the FDA or a comparable foreign regulatory authority does not
approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need
to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for
or market our product candidates, if approved.
We may have conflicts with our partners
that could delay or prevent the development or commercialization of our product candidates.
We may have conflicts with our
partners, such as conflicts concerning the interpretation of preclinical or clinical data, pertaining to the global patient safety profile
or efficacy results of our products, the achievement of milestones, the interpretation of contractual obligations, payments for services,
development obligations or the ownership of intellectual property developed during our collaboration. We may seek to amend, modify or
terminate agreements with partners, suppliers or service providers related to Iomab-B, Actimab-A or Iomab-ACT but there can be no assurance
that we can do so successfully or negotiate terms that are favorable to us. Failure of which can increase the risk of or result in litigation
or alternative dispute resolution options taken against us. Further, we may exercise our decision-making authority under certain circumstances
pertaining to global patient safety related to our products, which our partners may disagree with and may result in potential conflicts
and public disclosure of our rationale and position. If any conflicts arise with any of our partners, such partner may act in a manner
that is adverse to our best interests. Any such disagreement could result in one or more of the following, each of which could delay or
prevent the development or commercialization of our product candidates, and in turn prevent us from generating revenues: unwillingness
on the part of a partner to pay us milestone payments or royalties we believe are due under a collaboration; uncertainty regarding ownership
of intellectual property rights arising from our collaborative activities, which could prevent us from entering into additional collaborations;
unwillingness by the partner to cooperate in the development or manufacture of the product, including providing us with product data or
materials; unwillingness on the part of a partner to keep us informed regarding the progress of its development and commercialization
activities or to permit public disclosure of the results of those activities; initiating litigation or alternative dispute resolution
options by either party to resolve the dispute; or attempts by either party to terminate the agreement. Litigation or alternative
dispute resolution options can be lengthy and expensive, require significant time and attention from our management and are highly uncertain.
There can be no assurance that if we pursue, or a partner pursues litigation or alternative dispute resolution options, that we will prevail.
Monetary and equitable damages awarded against us could have a material adverse effect on our business.
If in the future we are unable to establish
U.S. or global sales and marketing capabilities or enter into agreements with third parties to sell and market our product candidates,
we may not be successful in commercializing our product candidates if they are approved and we may not be able to generate any revenue.
We currently do not have a
marketing or sales team for the marketing, sales and distribution of any of our product candidates that may receive regulatory approval.
In order to commercialize any product candidates after approval, we must build on a territory-by-territory basis marketing, sales, distribution,
managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and we may not be successful
in doing so. If our product candidates receive regulatory approval, we may decide to establish an internal sales or marketing team with
technical expertise and supporting distribution capabilities to commercialize our product candidates, which will be expensive and time-consuming
and will require significant attention of our executive officers to manage. Any failure or delay in the development of our internal sales,
marketing and distribution capabilities would adversely impact the commercialization of any of our product candidates that we obtain approval
to market.
With respect to the commercialization of all or certain of our product
candidates, we may choose to collaborate, either globally or on a territory-by-territory basis, with third parties that have direct sales
forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales
force and distribution systems. If we are unable to enter into or maintain such arrangements when needed on acceptable terms, or at all,
we may not be able to successfully commercialize any of our product candidates that receive regulatory approval or any such commercialization
may experience delays or limitations. If we are not successful in commercializing our product candidates, either on our own or through
collaborations with one or more third parties, our future product revenue will suffer and we may incur significant additional losses.
We face significant competition from other
biotechnology and pharmaceutical companies.
Our product candidates face,
and will continue to face, intense competition from large pharmaceutical and biotechnology companies, as well as academic and research
institutions. We compete in an industry that is characterized by (i) rapid technological change, (ii) evolving industry standards, (iii)
emerging competition and (iv) new product introductions. Our competitors have existing products and technologies that will compete with
our product candidates and technologies and may develop and commercialize additional products and technologies that will compete with
our product candidates and technologies. Because several competing companies and institutions have greater financial resources than us,
they may be able to (i) provide broader services and product lines, (ii) make greater investments in research and development, or R&D,
and (iii) carry on broader R&D initiatives. Our competitors also have greater development capabilities than we do and have substantially
greater experience in undertaking preclinical and clinical testing of product candidates, obtaining regulatory approvals, and manufacturing
and marketing pharmaceutical products. They also have greater name recognition and better access to customers than us.
Our product candidates may cause undesirable
side effects or have other properties that could halt their clinical development, prevent their regulatory approval, limit their commercial
potential, or result in significant negative consequences.
Undesirable side effects caused
by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more
restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign authorities. The drug-related side
effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability
claims. Any of these occurrences may harm our business, financial condition and prospects significantly. Even if any of our product candidates
receives marketing approval, as greater numbers of patients use a product following its approval, an increase in the incidence of side
effects or the incidence of other post-approval problems that were not seen or anticipated during pre-approval clinical trials could result
in a number of potentially significant negative consequences, including:
| |
● |
regulatory authorities may withdraw their approval of the product; |
| |
● |
regulatory authorities may require the addition of labeling statements, such as warnings or contraindications; |
| |
● |
we may be required to change the way the product is administered, conduct additional clinical trials or change the labeling of the product; |
| |
● |
we may elect, or we may be required, to recall or withdraw product from the market; |
| |
● |
we could be sued and held liable for harm caused to patients; and |
| |
● |
our reputation may suffer. |
Any of these events could
substantially increase the costs and expenses of developing, commercializing and marketing any such product candidates or could harm or
prevent sales of any approved products.
Risks Related to Our Intellectual Property
We depend upon securing and protecting critical
intellectual property.
We are dependent on obtaining
and maintaining patents, trade secrets, copyright and trademark protection of our technologies in the United States and other jurisdictions,
as well as successfully enforcing this intellectual property and defending this intellectual property against third-party challenges.
The degree of future protection of our proprietary rights is uncertain for product candidates that are currently in the early stages of
development because we cannot predict which of these product candidates will ultimately reach the commercial market or whether the commercial
versions of these product candidates will incorporate proprietary technologies.
Our patent position is highly uncertain
and involves complex legal and factual questions.
Accordingly, we cannot predict
the breadth of claims that may be allowed or enforced under our patents or in third-party patents. For example, we or our licensors might
not have been the first to make the inventions covered by each of our pending patent applications and issued patents; we or our licensors
might not have been the first to file patent applications for these inventions; others may independently develop similar or alternative
technologies or duplicate any of our technologies; it is possible that none of our pending patent applications or the pending patent applications
of our licensors will result in issued patents; our issued patents and issued patents of our licensors may not provide a basis for commercially
viable technologies, or may not provide us with any competitive advantages, or may be challenged and invalidated by third parties; and,
we may not develop additional proprietary technologies that are patentable.
Furthermore, the issuance
of a patent, while presumed valid and enforceable, is not conclusive as to its validity or its enforceability and it may not provide us
with adequate proprietary protection or competitive advantages against competitors with similar products. Competitors may also be able
to design around our patents. Other parties may develop and obtain patent protection for more effective technologies, designs or methods.
We may not be able to prevent the unauthorized disclosure or use of our technical knowledge or trade secrets by consultants, vendors,
former employees and current employees.
Patent rights are territorial,
and patent protection extends only to those countries where we have issued patents. Filing, prosecuting and defending patents on our products
and product candidates in all countries and jurisdictions throughout the world would be prohibitively expensive, and our intellectual
property rights in some countries outside the United States could be less extensive than those in the United States. Many countries, however,
do not protect intellectual property to the same extent as the U.S. or Europe, and their litigation processes differ. Competitors may
successfully challenge or avoid our patents, or manufacture products in countries where we have not applied for patent protection. Changes
in the patent laws in the U.S. or other countries may diminish the value of our patent rights. As a result of these and other factors,
the scope, validity, enforceability, and commercial value of our patent rights are uncertain and unpredictable.
Indeed, several companies
have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems
of some countries do not favor the enforcement of patents and other intellectual property rights, which could make it difficult for us
to stop the infringement, misappropriation or other violation of our intellectual property rights generally. Proceedings to enforce our
intellectual property rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other
aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk
of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that are initiated, and
the damages or other remedies awarded, if any, may not be commercially meaningful.
The patent positions of pharmaceutical
companies, including our patent position, involve complex legal and factual questions, and, therefore, the issuance, scope, validity and
enforceability of any patent claims that we may obtain cannot be predicted with certainty. Patents, if issued, may be challenged, deemed
unenforceable, invalidated, or circumvented. A third-party may submit prior art, or we may become involved in opposition, derivation,
reexamination, inter partes review, post-grant review, supplemental examination, or interference proceedings challenging our patent rights
or the patent rights of our licensors or development partners. The costs of defending or enforcing our proprietary rights in these proceedings
can be substantial, and the outcome can be uncertain. An adverse determination in any such submission or proceeding could reduce the scope
of, or invalidate, our patent rights, allow third parties to commercialize our technology or products and compete directly with us, or
reduce our ability to manufacture or commercialize products. Furthermore, if the scope or strength of protection provided by our patents
and patent applications is threatened, it could discourage companies from collaborating with us to license, develop or commercialize current
or future products. The ownership of our proprietary rights could also be challenged.
As a result, our owned and
licensed patents may not be valid, and we may not be able to obtain and enforce patents and to maintain trade secret protection for the
full commercial extent of our technology. The extent to which we are unable to do so could materially harm our business.
We or our licensors have applied
for and will continue to apply for patents for certain products and methods. Such applications may not result in the issuance of any patents,
and any patents now held or that may be issued may not provide us with adequate protection from competition. Furthermore, it is possible
that patents issued or licensed to us may be challenged successfully. In that event, if we have a preferred competitive position because
of such patents, such preferred position would be lost. If we are unable to secure or to continue to maintain a preferred position, we
could become subject to competition from the sale of generic products. Failure to receive, inability to protect, or expiration of our
patents for medical use, manufacture, conjugation and labeling of Ac-225, the antibodies that we license from third parties, or subsequent
related filings, would adversely affect our business and operations.
Patents issued or licensed
to us may be infringed by the products or processes of others. Our ability to enforce our patent rights depends on our ability to detect
infringement. It is difficult to detect infringers who do not advertise the components that are used in their products. Moreover, it may
be difficult or impossible to obtain evidence of infringement in a competitor’s or potential competitor’s product, particularly
in litigation in countries other than the U.S. that do not provide an extensive discovery procedure. Any litigation to enforce or defend
our patent rights, if any, even if we were to prevail, could be costly and time-consuming and would divert the attention of our management
and key personnel from our business operations. We may not prevail in any lawsuits that we initiate and the damages or other remedies
awarded if we were to prevail may not be commercially meaningful.
The cost of enforcing our
patent rights against infringers, if such enforcement is required, could be significant, and we may not have the financial resources to
fund such litigation. Further, such litigation can go on for years and the time demands could interfere with our normal operations. There
has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical industry.
We may become a party to patent litigation and other proceedings. The cost to us of any patent litigation, even if resolved in our favor,
could be substantial. Some of our competitors may be able to sustain the costs of such litigation more effectively than we can because
of their substantially greater financial resources. Litigation may also absorb significant management time.
Unpatented trade secrets,
improvements, confidential know-how and continuing technological innovation are important to our scientific and commercial success. Although
we attempt to and will continue to attempt to protect our proprietary information through reliance on trade secret laws and the use of
confidentiality agreements with our partners, collaborators, employees and consultants and other appropriate means, these measures may
not effectively prevent disclosure of our proprietary information, and, in any event, others may develop independently, or obtain access
to, the same or similar information. In addition, we cannot guarantee that we have executed these agreements with each party that may
have or have had access to our trade secrets. Furthermore, if the employees and consultants who are parties to these agreements breach
or violate the terms of these agreements, we may not have adequate remedies for any such breach or violation, and we could lose our trade
secrets through such breaches or violations.
Certain of our patent rights
are licensed to us by third parties. If we fail to comply with the terms of these license agreements, our rights to those patents may
be terminated, and we may be unable to conduct our business.
If we are found to be infringing patents
or trade secrets owned by others, we may be forced to cease or alter our product development efforts, obtain a license to continue the
development or sale of our products, and/or pay damages.
We may not have identified
all patents, published applications or published literature that affect our business either by blocking our ability to commercialize our
products, by preventing the patentability of one or more aspects of our products to us or our licensors, or by covering the same or similar
technologies that may affect our ability to market our products. For example, we (or our licensors) may not have conducted a patent clearance
search sufficient to identify potentially obstructing third party patent rights. Moreover, patent applications in the United States are
maintained in confidence for up to 18 months after their filing. In some cases, however, patent applications remain confidential in the
U.S. Patent and Trademark Office, or the USPTO, for the entire time prior to issuance as a U.S. patent. Patent applications filed in countries
outside of the United States are not typically published until at least 18 months from their first filing date. Similarly, publication
of discoveries in the scientific or patent literature often lags behind actual discoveries. We cannot be certain that we or our licensors
were the first to invent, or the first to file, patent applications covering our products and candidates. We also may not know if our
competitors filed patent applications for technology covered by our pending applications or if we were the first to invent the technology
that is the subject of our patent applications. Competitors may have filed patent applications or received patents and may obtain additional
patents and proprietary rights that block or compete with our patents.
Our manufacturing processes
and potential products may violate proprietary rights of patents that have been or may be granted to competitors, universities or others,
or the trade secrets of those persons and entities. As the pharmaceutical industry expands and more patents are issued, the risk increases
that our processes and potential products may give rise to claims that they infringe the patents or trade secrets of others. These other
persons could bring legal actions against us claiming damages and seeking to enjoin clinical testing, manufacturing and marketing of the
affected product or process. If any of these actions are successful, in addition to any potential liability for damages, we could be required
to obtain a license in order to continue to conduct clinical tests, manufacture or market the affected product or use the affected process.
Required licenses may not be available on acceptable terms, if at all, and the results of litigation are uncertain. If we become involved
in litigation or other proceedings, it could consume a substantial portion of our financial resources and the efforts of our personnel.
In addition to infringement
or other intellectual property claims against us, we may become a party to other patent litigation or proceedings before regulatory agencies,
including post-grant review, inter parties review, interference or re-examination proceedings filed with the U.S. Patent and Trademark
Office (or similar proceedings before corresponding tribunals in other jurisdictions) that challenge our patent rights or the patent rights
of our licensors. The costs and efforts of defending our patents or enforcing our proprietary rights in post-issuance administrative proceedings
can be substantial and the outcome can be uncertain. An adverse determination in these proceedings could weaken or invalidate the patent
claims that cover our technology, which adverse determination could harm our business significantly and dissuade companies from collaborating
with us or permit third parties to directly compete with the same technology.
Our ability to protect and enforce our patents
does not guarantee that we will secure the right to commercialize our potential products and respective patents.
A patent is a limited monopoly
right conferred upon an inventor, and his successors in title, in return for the making and disclosing of a new and non-obvious invention.
This monopoly is of limited duration but, while in force, allows the patent holder to prevent others from making, using and/or selling
its invention. While a patent gives the holder this right to exclude others, it is not a license to commercialize an invention covered
by the patent where other permissions may be required for commercialization to occur. For example, a drug cannot be marketed without the
appropriate authorization from the FDA, regardless of the existence of a patent covering the product. Further, the invention, even if
patented itself, cannot be commercialized if it infringes the valid patent rights of another party.
We rely on confidentiality agreements to
protect our trade secrets. If these agreements are breached by our employees or other parties, our trade secrets may become known to our
competitors.
We rely on trade secrets that
we seek to protect through numerous measures, including non-compete and confidentiality agreements with our employees and other parties.
If these agreements are breached, our competitors may obtain and use our trade secrets to gain a competitive advantage over us. Any remedies
that may be available to us may not be adequate to protect our business or compensate us for the damaging disclosure. In addition, we
may have to expend resources to protect our interests from possible infringement by others. For instance, we learned that a former employee,
Qing Liang, Ph.D., who was employed by Actinium in the position of Vice President, Head of Radiation Sciences, violated the non-compete
provision of her employment agreement by working for a direct competitor. Additionally, while working for the direct competitor, Dr. Liang
continued to provide consulting services to Actinium. We also learned that Dr. Liang was providing consulting services to another company,
which was in violation of certain provisions of her post-employment consulting agreement with Actinium. Dr. Liang, who had access to materials
containing proprietary information and trade secrets, pursuant to actions taken by Actinium, is no longer employed by the direct competitor.
With the assistance of outside counsel and a forensic investigator, we identified that Dr. Liang downloaded confidential information prior
to her employment at Actinium ending. To aid in arbitration proceedings, we petitioned and were granted a Stipulated Preliminary Injunction
by the Supreme Court of the State of New York, New York County (Index No. 656841/2022) on June 28, 2022 that ordered that Dr. Liang is
enjoined from destroying or deleting any Actinium documents or information, is enjoined from using, transmitting or transferring any Actinium
Information other than to her counsel or Actinium’s counsel, ordered to return Actinium information within 5 days of Stipulated
Preliminary Injunction, ordered to disclose to Actinium under oath, all persons and devices she transferred or disclosed Actinium Information,
and ordered to allow a qualified forensic examiner selected by Actinium to remove and permanently delete all Actinium Information from
any electronic devices, systems, email accounts, or other electronic or physical storage sites belonging to Dr. Liang. On April 25, 2023,
a Final Award and Permanent Injunction was granted by the Supreme Court of the State of New York, New York County (Case No. 01-22-0003-2375)
that ordered that Dr. Liang is permanently enjoined from using, possessing, transmitting or transferring any Actinium property, documents
of business information.
We may be subject to damages resulting from
claims that we or our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
Our employees may have been
previously employed at other companies in the industry, including our competitors or potential competitors. Although we are not aware
of any claims currently pending against us, we may be subject to claims that these employees or we have inadvertently or otherwise used
or disclosed trade secrets or other proprietary information of the former employers of our employees. Litigation may be necessary to defend
against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be
a distraction to management. If we fail in defending such claims, in addition to paying money claims, we may lose valuable intellectual
property rights or personnel. A loss of key personnel or their work product could hamper or prevent our ability to commercialize product(s),
which would materially adversely affect our commercial development efforts.
Obtaining and maintaining patent protection
depends on compliance with various procedures and other requirements, and our patent protection could be reduced or eliminated in case
of non-compliance with these requirements.
Periodic maintenance fees,
renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to the relevant patent agencies
in several stages over the lifetime of the patents and /or applications. The relevant patent agencies require compliance with a number
of procedural, documentary, fee payment and other provisions during the patent application process. In many cases, an inadvertent lapse
can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which
the failure to comply with the relevant requirements can result in the abandonment or lapse of the patent or patent application, resulting
in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to use our
technologies and know-how which could have a material adverse effect on our business, prospects, financial condition and results of operation.
Risks Related to Our Operations
We are highly dependent on our key personnel,
and if we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our
business strategy.
Our future operations and
successes depend in large part upon the continued service of key members of our senior management team whom we are highly dependent upon
to manage our business. If any member of our current senior management terminates his or her employment with us and we are unable to find
a suitable replacement quickly, the departure could have a material adverse effect on our business.
In the third quarter of 2024,
our overall headcount was reduced by approximately twenty percent, with a majority of departures coming from our clinical and CMC groups.
As a result of these departures, we expect our personnel expenses to be reduced by approximately $3.7 million in 2025, which may be offset
by additional hires or consultants. We do not expect these departures to have a material impact on our operations or ability to execute
our operating plan and are actively seeking a strategic partner for Iomab-B in the U.S. to advance the additional studies and trials required
by the FDA.
An overall tightening and
increasingly competitive labor market has been observed in the U.S. employment market generally. Specific to the biotechnology industry
in which we operate, there is significant demand and competition for highly specialized talent that we require. A sustained labor shortage
or increased turnover rates within our employee base as a result of general macroeconomic factors of force majeure events, or due
to dynamics within our industry, could lead to increased costs, such as increased wage rates to attract and retain employees, and could
negatively affect our ability to efficiently conduct our clinical development, R&D, business development and potential regulatory
and commercial activities. If we are unable to hire and retain employees capable of performing at a high-level, or if mitigation measures
we may take to respond to a decrease in labor availability, have unintended negative effects, our business could be adversely affected.
An overall labor shortage, lack of skilled labor, increased turnover or labor inflation, general macroeconomic factors or as a result
of biotechnology industry dynamics could have a material adverse impact on our operations, results of operations, liquidity or cash flows.
Our future success also depends
on our ability to identify, attract, hire, or engage, retain, and motivate other well-qualified managerial, technical, clinical and regulatory
personnel. This activity is likely to create additional demands on the time and attention of our senior management personnel as they identify,
hire, and train external and internal candidates to fill the sizable number of positions required to execute our business plans, including
submitting a BLA and building a commercial organization. The market for talent in our industry is very competitive. Many of the other
biopharmaceutical companies we compete against for qualified personnel have greater financial and other resources, more favorable risk
profiles and a longer operating history in the biopharmaceutical industry than we do. They also may provide more diverse opportunities
and better chances for career advancement. Some of these opportunities may be more appealing to high-quality candidates than what we have
to offer. There can be no assurance that such professionals will be available in the market, or that we will be able to retain existing
professionals or meet or continue to meet their compensation requirements. Furthermore, the cost base in relation to such compensation,
which may include equity compensation, may increase significantly, which could have a material adverse effect on us. Failure to establish
and maintain an effective management team and workforce could adversely affect our ability to operate, grow and manage our business.
Managing our growth as we expand operations
may strain our resources.
We expect to need to grow
rapidly in order to support additional, larger, and potentially international, pivotal clinical trials of our product candidates as well
as potential commercial operations in the future, which will place a significant strain on our financial, managerial and operational resources.
In order to achieve and manage growth effectively, we must continue to improve and expand our operational and financial management capabilities.
Moreover, we will need to increase staffing and to train, motivate and manage our employees. All of these activities will increase our
expenses and may require us to raise additional capital sooner than expected. Failure to manage growth effectively could materially harm
our business, financial condition or results of operations.
The use of hazardous materials, including
radioactive and biological materials, in our research and development efforts imposes certain compliance costs on us and may subject us
to liability for claims arising from the use or misuse of these materials.
Our research, development
and manufacturing activities involve the controlled use of hazardous materials, including chemicals, radioactive and biological materials,
such as radioactive isotopes. We are subject to federal, state, local and foreign environmental laws and regulations governing, among
other matters, the handling, storage, use and disposal of these materials and some waste products. We cannot completely eliminate the
risk of contamination or injury from these materials, and we could be held liable for any damages that result, which could exceed our
financial resources. We currently maintain insurance coverage for injuries resulting from the hazardous materials we use; however, future
claims may exceed the amount of our coverage. Also, we do not have insurance coverage for pollution cleanup and removal. Currently the
costs of complying with such federal, state, local and foreign environmental regulations are not significant, and consist primarily of
waste disposal expenses. However, they could become expensive, and current or future environmental laws or regulations may impair our
research, development, production and commercialization efforts.
We may undertake international operations,
which will subject us to risks inherent with operations outside of the United States.
Although we do not have any
international operations at this time, we intend to seek market clearances in foreign markets that we believe will generate significant
opportunities. However, even with the cooperation of a commercialization partner, conducting drug development in foreign countries involves
inherent risks, including, but not limited to difficulties in staffing, funding and managing foreign operations; unexpected changes in
regulatory requirements; export restrictions; tariffs and other trade barriers; difficulties in protecting, acquiring, enforcing and litigating
intellectual property rights; fluctuations in currency exchange rates; and potentially adverse tax consequences.
If we were to experience any
of the difficulties listed above, or any other difficulties, any international development activities and our overall financial condition
may suffer and cause us to reduce or discontinue our international development and registration efforts.
We expect to expand our development and
regulatory capabilities and potentially implement sales, marketing and distribution capabilities, and, as a result, we may encounter difficulties
in managing our growth, which could disrupt our operations.
We expect to experience growth
in the number of our employees and the scope of our operations, particularly in the areas of product candidate development, regulatory
affairs and, if any of our product candidates receives marketing approval, sales, marketing, and distribution.
We currently do not have a
marketing or sales team for the marketing, sales and distribution of any of our product candidates that are potentially able to obtain
regulatory approval. In order to commercialize any product candidates, we must build on a territory-by-territory basis marketing, sales,
distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and we
may not be successful in doing so. If our product candidates receive regulatory approval, we intend to establish an internal sales or
marketing team with technical expertise and supporting distribution capabilities to commercialize our product candidates, which will be
expensive and time consuming and will require significant attention of our executive officers to manage. We will also have to compete
with other pharmaceutical and biotechnology companies to recruit, hire, train and retain marketing and sales personnel. Any failure or
delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of
any of our product candidates that we obtain approval to market.
To manage our anticipated
future growth, we must continue to implement and improve our managerial, operational, and financial systems, expand our facilities, and
continue to recruit and train additional qualified personnel. Due to our limited financial resources and the limited experience of our
management team in managing a public company with such anticipated growth, we may not be able to effectively manage the expansion of our
operations or recruit and train additional qualified personnel. The expansion of our operations may lead to significant costs and may
divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans
or disrupt our operations.
We continuously evaluate our business strategy
and may modify our strategy as necessary to respond to developments in our business and other factors, and any such modification, if not
successful, could have a material adverse effect on our business, financial condition, and results of operations.
We continuously evaluate our
business strategy and modify our plans as necessary to achieve our objectives in response to changing circumstances. As part of such a
process, we may delay, modify or discontinue the development of certain of our drug candidates and choose alternative approaches if we
believe such changes would be in our best interest. We may also expand or alter our research and development activities from time to time
and redirect allocation of our resources. We have implemented such changes in our business strategy and may continue to do so in the future.
There can be no assurances that any product development or other changes that we implement will be successful or that, after implementation
of any such changes, that we will not refocus our efforts on new or different objectives.
We may expand our business through the acquisition
of rights to new product candidates that could disrupt our business, harm our financial condition and may also dilute current stockholders’
ownership interests in our company.
Our business strategy includes
expanding our products and capabilities, and we may seek acquisitions of product candidates, antibodies or technologies to do so. Acquisitions
involve numerous risks, including substantial cash expenditures; potentially dilutive issuance of equity securities; incurrence of debt
and contingent liabilities, some of which may be difficult or impossible to identify at the time of acquisition; difficulties in assimilating
acquired technologies or the operations of the acquired companies; diverting our management’s attention away from other business
concerns; risks of entering markets in which we have limited or no direct experience; and the potential loss of our key employees or key
employees of the acquired companies.
We can make no assurances
that any acquisition will result in short-term or long-term benefits to us. We may incorrectly judge the value or worth of an acquired
product, company or business. In addition, our future success would depend in part on our ability to manage the rapid growth associated
with some of these acquisitions. We cannot assure that we will be able to make the combination of our business with that of acquired products,
businesses or companies work or be successful. Furthermore, the development or expansion of our business or any acquired products, business
or companies may require a substantial capital investment by us. We may not have these necessary funds, or they might not be available
to us on acceptable terms or at all. We may also seek to raise funds by selling shares of our preferred or common stock, which could dilute
each current stockholder’s ownership interest in the Company.
Risks Related to Ownership of Our Common Stock
The sale of securities by us in any equity
or debt financing could result in dilution to our existing stockholders and have a material adverse effect on our earnings.
We have financed our operations
primarily through sales of stock and warrants. It is likely that during the next twelve months we will seek to raise additional capital
through the sales of stock and warrants in order to expand our level of operations to continue our research and development efforts.
Any sale of common stock by
us in a future offering could result in dilution to our existing stockholders as a direct result of our issuance of additional shares
of our capital stock. In addition, our business strategy may include expansion through internal growth or by establishing strategic relationships
with targeted customers and vendors. In order to do so, or to finance the cost of our other activities, we may issue additional equity
securities that could dilute our stockholders’ stock ownership. We may also assume additional debt and incur impairment losses related
to goodwill and other tangible assets if we acquire another company and this could negatively impact our earnings and results of operations.
Our common stock is subject to price volatility which could
lead to losses by stockholders and potential costly security litigation.
The trading volume of our
common stock has been and may continue to be extremely limited and sporadic. We expect the market price of our common stock to fluctuate
substantially due to a variety of factors, including market perception of our ability to achieve our planned growth, quarterly operating
results of other companies in the same industry, trading volume in our common stock, changes in general conditions in the economy and
the financial markets or other developments affecting our competitors or us. This volatility has had a significant effect on the market
price of securities issued by many companies for reasons unrelated to their operating performance and could have the same effect on our
common stock.
The trading price of our common
stock may be highly volatile and could fluctuate in response to factors such as:
| |
● |
actual or anticipated variations in our operating results; |
| |
● |
announcements of developments by us or our competitors; |
| |
● |
the timing of IND and/or BLA approval, the completion and/or results of our clinical trials; |
| |
● |
regulatory actions regarding our products; |
| |
● |
announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments; |
| |
● |
adoption of new accounting standards affecting our industry; |
| |
● |
additions or departures of key personnel; |
| |
● |
introduction of new products by us or our competitors; |
| |
● |
sales of our common stock or other securities in the open market; |
| |
● |
inaccurate or unfavorable reports from securities or industry analysts; and |
| |
● |
other events or factors, many of which are beyond our control. |
The stock market is subject
to significant price and volume fluctuations. In the past, following periods of volatility in the market price of a company’s securities,
securities class action litigation has often been initiated against such a company. Litigation initiated against us, whether or not successful,
could result in substantial costs and diversion of our management’s attention and our resources, which could harm our business and
financial condition.
We do not intend to pay dividends on our
common stock, so any returns will be determined by the value of our common stock.
We have never declared or
paid any cash dividends on our common stock. For the foreseeable future, it is expected that earnings, if any, generated from our operations
will be used to finance the growth of our business, and that no dividends will be paid to holders of our common stock. As a result, the
success of an investment in our common stock will depend upon any future appreciation in its value. There is no guarantee that our common
stock will appreciate in value.
Certain provisions of our Certificate of
Incorporation and Bylaws and Delaware law make it more difficult for a third party to acquire us and make a takeover more difficult to
complete, even if such a transaction were in our stockholders’ interest.
Provisions of our certificate
of incorporation and bylaws may delay or discourage transactions involving an actual or potential change in our control or change in our
management, including transactions in which stockholders might otherwise receive a premium for their shares, or transactions that our
stockholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our stock.
Among other things, the certificate of incorporation and bylaws:
| |
● |
provide that the authorized number of directors may be changed by resolution of the board of directors; |
| |
● |
provide that all vacancies, including newly-created directorships, may, except as otherwise required by law, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum; |
| |
● |
divide the board of directors into three classes; |
| |
● |
provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide notice in writing in a timely manner, and meet specific requirements as to the form and content of a stockholder’s notice; |
In addition, we are governed
by Section 203 of the Delaware General Corporation Law. In general, Section 203 prohibits a public Delaware corporation from engaging
in a “business combination” with an “interested stockholder” for a period of three years after the date of the
transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner.
A “business combination” includes mergers, asset sales or other transactions resulting in a financial benefit to the stockholder.
An “interested stockholder” is a person who, together with affiliates and associates, owns, or within three years, did own,
15% or more of the corporation’s outstanding voting stock. These provisions may have the effect of delaying, deferring or preventing
a change in our control.
General Risk Factors
Compliance with the reporting requirements
of federal securities laws can be expensive.
We are subject to the information
and reporting requirements of the Exchange Act and other federal securities laws, and the compliance obligations of the Sarbanes-Oxley
Act. The costs of preparing and filing annual and quarterly reports and other information with the Securities and Exchange Commission
and furnishing audited reports to stockholders are substantial. In addition, we will incur substantial expenses in connection with the
preparation of registration statements and related documents with respect to any offerings of our common stock.
Our ability to utilize our net operating
loss carryforwards and certain other tax attributes may be limited.
Our ability to utilize our
federal net operating loss and tax credit carryforwards may be limited under Sections 382 and 383 of the Internal Revenue Code of 1986,
as amended, or the Code. The limitations apply if we experience an “ownership change”, generally defined as a greater
than 50 percentage point change in the ownership of our equity by certain stockholders over a rolling three-year period. Similar
provisions of state tax law may also apply. We have not assessed whether such an ownership change has previously occurred. If we
have experienced an ownership change at any time since our formation, we may already be subject to limitations on our ability to utilize
our existing net operating losses and other tax attributes to offset taxable income. In addition, future changes in our stock ownership,
which may be outside of our control, may trigger an ownership change and, consequently, the limitations under Sections 382 and 383 of
the Code. As a result, if or when we earn net taxable income, our ability to use our pre-change net operating loss carryforwards
and other tax attributes to offset such taxable income may be subject to limitations, which could adversely affect our future cash flows.
Failure to establish and maintain adequate
finance infrastructure and accounting systems and controls could impair our ability to comply with the financial reporting and internal
controls requirements for publicly traded companies.
As a public company, we operate
in an increasingly demanding regulatory environment, including with respect to more complex accounting rules. Company responsibilities
required by the Sarbanes-Oxley Act of 2002, as amended, or the Sarbanes-Oxley Act, include establishing and maintaining corporate oversight
and adequate internal control over financial reporting and disclosure controls and procedures. Effective internal controls are necessary
for us to produce reliable financial reports and are important to help prevent financial fraud.
Our compliance with Section
404 of the Sarbanes-Oxley Act requires that we incur substantial accounting expense and expend significant management efforts. We complied
with Section 404 at December 31, 2023 and 2022 and while our testing did not reveal any material weaknesses in our internal controls,
any material weaknesses in our internal controls in the future would be required us to remediate in a timely manner so as to be able to
comply with the requirements of Section 404 each year. If we are not able to comply with the requirements of Section 404 in a timely manner
each year, we could be subject to sanctions or investigations by the SEC, NYSE American or other regulatory authorities which would require
additional financial and management resources and could adversely affect the market price of our common stock. Furthermore, if we cannot
provide reliable financial reports or prevent fraud, our business and results of operations could be harmed, and investors could lose
confidence in our reported financial information.
If securities or industry analysts do not
publish research or publish inaccurate or unfavorable research about our business, the price of our common stock and trading volume could
decline.
The trading market for our
common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. Multiple
securities and industry analysts currently cover us. If one or more of the analysts downgrade our common stock or publish inaccurate or
unfavorable research about our business, the price of our common stock would likely decline. If one or more of these analysts cease coverage
of us or fail to publish reports on us regularly, demand for our common stock could decrease, which could cause the price of our common
stock and trading volume to decline.
Our amended and restated bylaws, as
amended, designate the U.S. federal district courts as the exclusive forum for the resolution of any complaint
asserting a cause of action arising under the Securities Act of 1933, as amended.
Our amended and restated bylaws,
as amended, provide that, unless we consent in writing to the selection of an alternative forum, the federal district courts of the
United States of America will be the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities
Act of 1933, as amended. In addition, our amended and restated bylaws, as amended, state that any person purchasing or otherwise acquiring
any interest in our security shall be deemed to have notice of and to have consented to such provision. Such choice of forum provision
may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors,
officers or other employees, which may discourage such lawsuits, if successful, might benefit our stockholders. Stockholders who do bring
a claim in the federal district courts of the United States of America could face additional litigation costs in pursuing any such claim.
ITEM 2. UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS.
None.
ITEM 3. DEFAULTS UPON SENIOR SECURITIES.
None.
ITEM 4. MINE SAFETY DISCLOSURES.
None.
ITEM 5. OTHER INFORMATION.
ITEM 6. EXHIBITS
Copies of the following documents
are included as exhibits to this report pursuant to Item 601 of Regulation S-K.
| Exhibit No. |
|
Description |
| 3.1 |
|
Certificate of Incorporation of Actinium Pharmaceuticals, Inc. (incorporated by reference to Exhibit 3.1 of the Company’s Form 8-K filed with the SEC on April 17, 2013). |
| |
|
|
| 3.2 |
|
Certificate of Amendment to Certificate of Incorporation, as amended, filed January 7, 2014 (incorporated by reference to Exhibit 3.5 to Form S-1 filed on January 31, 2014). |
| |
|
|
| 3.3 |
|
Certificate of Amendment to Certificate of Incorporation, as amended, filed February 3, 2014. (incorporated by reference to Exhibit 3.1 to Form 8-K filed on February 7, 2014). |
| |
|
|
| 3.4 |
|
Certificate of Amendment to Certificate of Incorporation, as amended, filed on February 26, 2015 (incorporated by reference to Exhibit 3.1 to Form 8-K filed on March 4, 2015). |
| |
|
|
| 3.5 |
|
Certificate of Amendment to Certificate of Incorporation, as amended, filed on February 26, 2018 (incorporated by reference to Exhibit 3.1 to Form 8-K filed on February 26, 2018). |
| |
|
|
| 3.6 |
|
Certificate of Amendment to Certificate of Incorporation, as amended, filed on March 6, 2019 (incorporated by reference to Exhibit 3.7 to Form 10-K filed on March 15, 2019). |
| |
|
|
| 3.7 |
|
Certificate of Amendment to Certificate of Incorporation, as amended, filed on June 16, 2020 (incorporated by reference to Exhibit 3.1 to Form 8-K filed on June 16, 2020). |
| |
|
|
| 3.8 |
|
Certificate of Amendment to Certificate of Incorporation, as amended, filed on August 10, 2020 (incorporated by reference to Exhibit 3.1 to Form 8-K filed on August 14, 2020). |
| |
|
|
| 3.9 |
|
Amended and Restated Bylaws, dated August 9, 2018 (incorporated by reference to Exhibit 3.1 to Form 10-Q filed on July 26, 2018). |
| |
|
|
| 3.10 |
|
Amendment to Amended and Restated Bylaws, dated May 7, 2020 (incorporated by reference to Exhibit 3.1 to Form 8-K filed on May 5, 2020). |
| |
|
|
| 31.1* |
|
Certification of the Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
| |
|
|
| 31.2* |
|
Certification of the Principal Financial and Accounting Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
| |
|
|
| 32.1** |
|
Certification of the Chief Executive Officer pursuant to U.S.C. Section 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002* |
| |
|
|
| 32.2** |
|
Certification of the Principal Financial and Accounting Officer pursuant to U.S.C. Section 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002* |
| |
|
|
| 101.INS* |
|
Inline XBRL Instance Document |
| 101.SCH* |
|
Inline XBRL Taxonomy Schema Document |
| 101.CAL* |
|
Inline XBRL Taxonomy Calculation Linkbase Document |
| 101.DEF* |
|
Inline XBRL Taxonomy Definition Linkbase Document |
| 101.LAB* |
|
Inline XBRL Taxonomy Label Linkbase Document |
| 101.PRE* |
|
Inline XBRL Taxonomy Presentation Linkbase Document |
| 104* |
|
Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101). |
SIGNATURES
Pursuant to the requirements
of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto
duly authorized.
| |
ACTINIUM PHARMACEUTICALS, INC. |
| |
|
|
| Date: November 14, 2024 |
By: |
/s/ Sandesh Seth |
| |
|
Sandesh Seth |
| |
|
Chairman and Chief Executive Officer
(Duly Authorized Officer and
Principal Executive Officer) |
| |
|
|
| |
By: |
/s/ Steve O’Loughlin |
| |
|
Steve O’Loughlin |
| |
|
Chief Financial Officer |
| |
|
(Duly Authorized Officer and
Principal Financial and Accounting Officer) |
62
false
--12-31
Q3
0001388320
0001388320
2024-01-01
2024-09-30
0001388320
2024-11-14
0001388320
2024-09-30
0001388320
2023-12-31
0001388320
atnm:RevenueMember
2024-07-01
2024-09-30
0001388320
atnm:RevenueMember
2023-07-01
2023-09-30
0001388320
atnm:RevenueMember
2024-01-01
2024-09-30
0001388320
atnm:RevenueMember
2023-01-01
2023-09-30
0001388320
atnm:OtherRevenueMember
2024-07-01
2024-09-30
0001388320
atnm:OtherRevenueMember
2023-07-01
2023-09-30
0001388320
atnm:OtherRevenueMember
2024-01-01
2024-09-30
0001388320
atnm:OtherRevenueMember
2023-01-01
2023-09-30
0001388320
2024-07-01
2024-09-30
0001388320
2023-07-01
2023-09-30
0001388320
2023-01-01
2023-09-30
0001388320
us-gaap:CommonStockMember
2023-12-31
0001388320
us-gaap:AdditionalPaidInCapitalMember
2023-12-31
0001388320
us-gaap:RetainedEarningsMember
2023-12-31
0001388320
us-gaap:CommonStockMember
2024-01-01
2024-03-31
0001388320
us-gaap:AdditionalPaidInCapitalMember
2024-01-01
2024-03-31
0001388320
us-gaap:RetainedEarningsMember
2024-01-01
2024-03-31
0001388320
2024-01-01
2024-03-31
0001388320
us-gaap:CommonStockMember
2024-03-31
0001388320
us-gaap:AdditionalPaidInCapitalMember
2024-03-31
0001388320
us-gaap:RetainedEarningsMember
2024-03-31
0001388320
2024-03-31
0001388320
us-gaap:CommonStockMember
2024-04-01
2024-06-30
0001388320
us-gaap:AdditionalPaidInCapitalMember
2024-04-01
2024-06-30
0001388320
us-gaap:RetainedEarningsMember
2024-04-01
2024-06-30
0001388320
2024-04-01
2024-06-30
0001388320
us-gaap:CommonStockMember
2024-06-30
0001388320
us-gaap:AdditionalPaidInCapitalMember
2024-06-30
0001388320
us-gaap:RetainedEarningsMember
2024-06-30
0001388320
2024-06-30
0001388320
us-gaap:CommonStockMember
2024-07-01
2024-09-30
0001388320
us-gaap:AdditionalPaidInCapitalMember
2024-07-01
2024-09-30
0001388320
us-gaap:RetainedEarningsMember
2024-07-01
2024-09-30
0001388320
us-gaap:CommonStockMember
2024-09-30
0001388320
us-gaap:AdditionalPaidInCapitalMember
2024-09-30
0001388320
us-gaap:RetainedEarningsMember
2024-09-30
0001388320
us-gaap:CommonStockMember
2022-12-31
0001388320
us-gaap:AdditionalPaidInCapitalMember
2022-12-31
0001388320
us-gaap:RetainedEarningsMember
2022-12-31
0001388320
2022-12-31
0001388320
us-gaap:CommonStockMember
2023-01-01
2023-03-31
0001388320
us-gaap:AdditionalPaidInCapitalMember
2023-01-01
2023-03-31
0001388320
us-gaap:RetainedEarningsMember
2023-01-01
2023-03-31
0001388320
2023-01-01
2023-03-31
0001388320
us-gaap:CommonStockMember
2023-03-31
0001388320
us-gaap:AdditionalPaidInCapitalMember
2023-03-31
0001388320
us-gaap:RetainedEarningsMember
2023-03-31
0001388320
2023-03-31
0001388320
us-gaap:CommonStockMember
2023-04-01
2023-06-30
0001388320
us-gaap:AdditionalPaidInCapitalMember
2023-04-01
2023-06-30
0001388320
us-gaap:RetainedEarningsMember
2023-04-01
2023-06-30
0001388320
2023-04-01
2023-06-30
0001388320
us-gaap:CommonStockMember
2023-06-30
0001388320
us-gaap:AdditionalPaidInCapitalMember
2023-06-30
0001388320
us-gaap:RetainedEarningsMember
2023-06-30
0001388320
2023-06-30
0001388320
us-gaap:CommonStockMember
2023-07-01
2023-09-30
0001388320
us-gaap:AdditionalPaidInCapitalMember
2023-07-01
2023-09-30
0001388320
us-gaap:RetainedEarningsMember
2023-07-01
2023-09-30
0001388320
us-gaap:CommonStockMember
2023-09-30
0001388320
us-gaap:AdditionalPaidInCapitalMember
2023-09-30
0001388320
us-gaap:RetainedEarningsMember
2023-09-30
0001388320
2023-09-30
0001388320
us-gaap:EmployeeStockOptionMember
2024-01-01
2024-09-30
0001388320
us-gaap:EmployeeStockOptionMember
2023-01-01
2023-09-30
0001388320
us-gaap:RestrictedStockUnitsRSUMember
2024-01-01
2024-09-30
0001388320
us-gaap:RestrictedStockUnitsRSUMember
2023-01-01
2023-09-30
0001388320
us-gaap:WarrantMember
2024-01-01
2024-09-30
0001388320
us-gaap:WarrantMember
2023-01-01
2023-09-30
0001388320
2012-06-15
2012-06-15
0001388320
us-gaap:CorporateAndOtherMember
2023-12-31
0001388320
atnm:OperatingLeasesMember
2024-09-30
0001388320
atnm:FinanceLeasesMember
2024-09-30
0001388320
2022-04-07
0001388320
atnm:CapitalOnDemandSalesAgreementMember
2020-08-31
2020-08-31
0001388320
us-gaap:CommonStockMember
2024-01-01
2024-09-30
0001388320
us-gaap:CommonStockMember
2023-01-01
2023-09-30
0001388320
us-gaap:StockOptionMember
2024-01-01
2024-09-30
0001388320
srt:MinimumMember
us-gaap:StockOptionMember
2024-01-01
2024-09-30
0001388320
srt:MaximumMember
us-gaap:StockOptionMember
2024-01-01
2024-09-30
0001388320
srt:MinimumMember
2024-01-01
2024-09-30
0001388320
srt:MaximumMember
2024-01-01
2024-09-30
0001388320
us-gaap:StockOptionMember
2024-09-30
0001388320
us-gaap:StockOptionMember
2023-01-01
2023-09-30
0001388320
us-gaap:RestrictedStockUnitsRSUMember
2024-01-01
2024-09-30
0001388320
us-gaap:RestrictedStockUnitsRSUMember
2024-09-30
0001388320
2024-01-01
0001388320
2024-01-01
2024-01-01
0001388320
us-gaap:RestrictedStockUnitsRSUMember
2023-12-31
0001388320
us-gaap:WarrantMember
2024-01-01
0001388320
us-gaap:WarrantMember
2024-01-01
2024-01-01
0001388320
us-gaap:WarrantMember
2024-01-01
2024-09-30
0001388320
us-gaap:WarrantMember
2024-09-30
xbrli:shares
iso4217:USD
iso4217:USD
xbrli:shares
xbrli:pure
18 U.S.C. SECTION 1350,
18 U.S.C. SECTION 1350,
18 U.S.C. SECTION 1350,
In connection with the Quarterly Report of Actinium Pharmaceuticals,
Inc. (the “Company”) on Form 10-Q for the period ended September 30, 2024 as filed with the Securities and Exchange Commission
on the date hereof (the “Report”), I, Sandesh Seth, Chairman & CEO of the Company, certify, pursuant to 18 U.S.C. Section
1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that:
18 U.S.C. SECTION 1350,
In connection with the Quarterly Report of Actinium Pharmaceuticals,
Inc. (the “Company”) on Form 10-Q for the period ended September 30, 2024 as filed with the Securities and Exchange Commission
on the date hereof (the “Report”), I, Steve O’Loughlin, Chief Financial Officer of the Company, certify, pursuant to
18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that: